
GroEL. "Top view" C-alpha trace colored by crystrallographic B-factors. Red indicates high mobility; blue low mobility. The inside of the apical domains, where unfolded substrates are thought to bind, are the most mobile.
landry@mailhost.tcs.tulane.edu
GroEL. "Top view" C-alpha trace colored by crystrallographic
B-factors. Red indicates high mobility; blue low mobility. The inside of
the apical domains, where unfolded substrates are thought to bind, are
the most mobile.

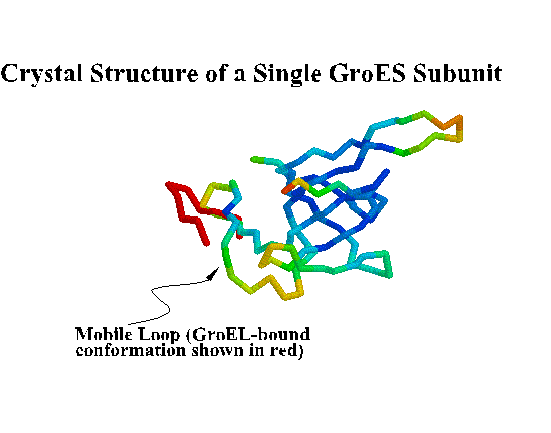
GroES. "Top view" C-alpha trace colored by crystallographic B-factors. Only one of seven mobile loops is visible at left. For a closer look examine the kinemage estour_2.kin. [If necessary, first get the kinemage viewer Mage 4.2 ]

GroES and GroEL. "Side view" showing the four-banded appearance of the GroEL "double donut" and dome-shaped GroES. Each GroEL donut is composed of a ring of seven subunits. Each subunit is composed of three domains: apical, intermediate, and equatorial. The equatorial domains are well ordered in the crystal, as indicated by low B-factors (blue). ATP binds to the equatorial domains triggering a conformational change in the intermediate and apical domains that leads to discharge of the folding protein. In addition, allosteric interactions are transmitted from one ring to the other. For example, when GroES binds to one end, binding of GroES to the other end becomes much less favorable. When GroES binds, its seven mobile loops contact the apical domains of GroEL. The one visible mobile loop is probably not in the GroEL-binding conformation, but rather must swing down to make contact with GroEL. The roof of the GroES dome is not very stable, and it is possible that some chaperonin substrates leave the GroEL/GroES right through a hole in the roof!
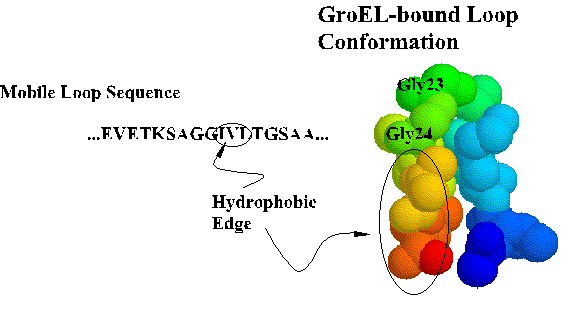
Coordinates for the trNOE NMR structure of the central nine residues of the GroES mobile loop are available from the Brookhaven Protein Database under entry 1EGS. This file contains twenty structures generated by molecular dynamics and simulated annealing. Structure #17 is shown below and in Landry et al., 1996. Recently, Xu et al. (1997) solved the X-ray crystal structure of the GroES-GroEL-ADP complex and found the loop in a beta hairpin conformation as we described.

The Chaperonin ATPase Cycle: Mechanism of Allosteric Switching, a movie published on the Web by the Saibil group and associated with the article by Alan M. Roseman, Shaoxia Chen, Helen White, Kerstin Braig, and Helen R. Saibil in the October 18, 1996 issue of Cell.
Landry, S. J., and Gierasch, L. M. (1994). Polypeptide Interactions with Molecular Chaperones and the Relationship to In Vivo Protein Folding. Ann. Rev. Biophys. Biomolec. Struct. 23, 645-669.
Todd, M. J., Viitanen, P. V., and Lorimer, G. H. (1994). Dynamics of the chaperonin ATPase cycle: Implications for facilitated protein folding. Science 265, 659-666.
Weissman, J. S., Kashi, Y., Fenton, W. A., and Horwich, A. L. (1994). GroEL-mediated protein folding proceeds by multiple rounds of binding and release of nonnative forms. Cell 78, 693-702.
Braig, K., Otwinowski, Z., Hegde, R., Boisvert, D. C., Joachimiak, A., Horwich, A. L., and Sigler, P. B. (1994). The crystal structure of the bacterial chaperonin GroEL at 2.8 angstrom. Nature 371, 578-586.
Fenton, W. A., Kashi, Y., Furtak, K., and Horwich, A. L. (1994). Residues in chaperonin GroEL required for polypeptide binding and release. Nature 371, 614-619.
Weissman, J. S., Hohl, C. M., Kovalenko, O., Kashi, Y., Chen, S. X., Braig, K., Saibil, H. R., Fenton, W. A., and Horwich, A. L. (1995). Mechanism of GroEL action: Productive release of polypeptide from a sequestered position under GroES. Cell 83, 577-587.
Hunt, J. F., Weaver, A. J., Landry, S. J., Gierasch, L., and Deisenhofer, J. (1996). The crystal structure of the GroES co-chaperonin at 2.8 angstrom resolution. Nature 379, 37-45.
Landry, S. J., Taher, A., Georgopoulos, C., and van der Vies, S. M. (1996). Interplay of structure and disorder in co-chaperonin mobile loops. Proc. Natl. Acad. Sci. U. S. A. 93, 11622-11627.
Saibil, H. R. (1996). Chaperonin structure and conformational changes. In Cell Biology, A Series of, Monographs: Chaperonins, R. J. Ellis, ed. (San Diego: Academic Press Inc), pp. 245-266.
Török, Z., Vigh, L., and Goloubinoff, P. (1996). Fluorescence detection of symmetric GroEL(14)(GroES(7))(2) heterooligomers involved in protein release during the chaperonin cycle. J. Biol. Chem. 271, 16180-16186.
Hartl, F. U. (1996). Molecular chaperones in cellular protein folding. Nature 381, 571-580.
Azem, A., Diamant, S., Kessel, M., Weiss, C., and Goloubinoff, P. (1995). The protein-folding activity of chaperonins correlates with the symmetric GroEL-14(GroES-7)-2 heterooligomer. Proc. Natl. Acad. Sci. U. S. A. 92, 12021-12025.
Xu, Z., Horwich, A. L., and Sigler, P. B. (1997). The crystal structure
of the asymmetric GroEL-GroES-(ADP)7 chaperonin
complex. Nature 388, 741-750.
End of document