Provides wealth of information about sequences being analyzed.
Structural information - protein alignment can reveal regions most conserved
and critical for function, i.e. active site residues
Chemical nature can be used to infer possible chemistry necessary for reaction.
Hidden Markov Model (HMM) sequence alignments can be used to train program to identify function of unknown ORFs
Less strongly conserved residues may reveal what characteristics are important for their structural role, i.e. conserved alternating pattern of hydrophilic and hydrophobic residues may indicate a beta sheet secondary structure.
Regions that have no insertions or deletions are likely to have specific structure, while regions of variable length and sequence might indicate variable loops or linkers between domains.
Conserved regions of nucleic acids may indicate important protein binding sites (transcriptional or translationsal control factors, for example)or active site residues of a ribozyme ;-)
Nucleic acid conserved regions reveal consensus sequence important for protein
binding
can also be used in designing PCR primers to amplify DNA from diverse species
Since conserved regions tend to cluster at sites across alignment, may find conserved sites for PCR primers to amplify non-conserved region between them.
Alignments can also be used to infer evolutionary relationships amongst the organisms from which they were obtained.
More related organisms tend to have more similar sequences for an orthologous protein or nucleic acid than would more distantly related ones.
Relative conservation of sequences can be used to construct a phylogenetic tree of the relatedness of those organisms.
Can be used to attempt to identify which variations in sequences occurred in which order.
Can also be used to identify residues in a sequence which appear to interact with other residues because their identities seem to have changed simultaneously with another to maintain function.
Called covariation. Particularly important in defining RNA structures. Covariation of Watson Crick base pairs is indicative of a double-helical RNA stem.
The same principle can be applied to paralogous sequences (duplicated genes
which have acquired new functions within an organism.) Can be used to infer
when the gene duplication arose and what residues were important for acquisition
of new function .
Information from multiple alignment can help to refine a pairwise alignment
of sequences. Remember the BLAST search exercises showed variations in alignments
, depending upon matrix used and gap penalties.
By looking at additional sequences, one might find intermediately related sequences
which help in optimizing alignment.
Or multiple alignment may reveal which regions appear most prone to gaps, and
so aid in aligning 2 particular sequences of interest
How can you tell if a multiple sequence alignment is meaningful?
Structural alignment is generally considered the standard for aligning molecules,
particularly proteins.
In a few cases there are crystal structures of the same protein from multiple
organisms with multiple sequences.
In this case, one can align the 3-D protein structures and identify residues
from the structures which occupy the same region of space. This allows unequivocal
alignment of the primary structures.
Usually, there are many more primary sequences available than tertiary structures,
so other methods must be used, but they can compared to test cases where structural
alignments are known. In general, sequence alignment programs can provide alignments
which agree well with structural alignments.
In theory, you can perform optimal alignment of multiple sequences by extension
of pairwise algorithms, but number of calculations needed is the sequence length
raised to the power of the number of sequences, so it is generally impractical
to calculate true optimal sequence alignment for more than 3 sequences.
One method to work around the computational problem is progressive alignment
based upon pairwise alignment of all sequences
CLUSTAL Has number of variations, the most commonly used is:
CLUSTALW
Hierarchical multiple alignment program.
Generates pairwise alignments of all input sequences, then ranks scores of identities
among pairs of sequences.
High scoring pairs of sequences align most readily to each other.
More divergent (less related) pairs are then added to the alignment.
So it generates a phylogenetic tree of relationships to determine steps in constructing
the alignment.
One can view the phylogenetic tree used to generate the alignment
Individual pairs in the alignment are aligned using a FASTA-type (word-based,
fast alignment) or by a dynamic programming algorithm, which is slower, but
produces optimal pairwise alignments.
|
FASTA-like algorithm |
|
Slower, optimal algorithm |
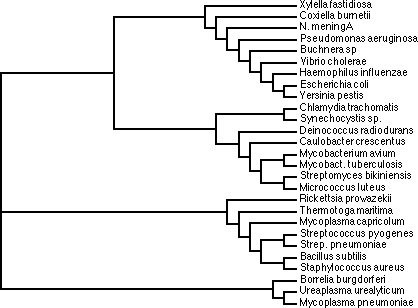
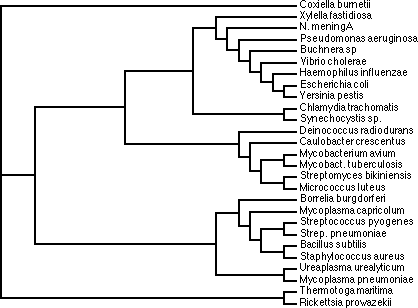
Trees based on alignment of RNaseP protein sequences. Note that some sequences
are grouped with different organisms depending upon algorithm.
Could have big effect on tree in some cases, but in this case it did not. Compare
the fast alignment pdf and the
slow alignment pdf
Note that in both cases the secondary structure is only interrupted once in
a helix (h) or sheet (s).
Dynamic Programming
Takes results of dot matrix and computes new matrix with penalties for gaps
Highest score at end of matrix represents optimal alignment end point.
|
|
|
||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|
|
Word Size = 3
|
|
Path is then traced back by shortest route to highest score diagonally above
Result is optimal pairwise alignment
Used for both local (Smith-Waterman) and global (Needleman-Wunsch) pairwise alignments
Order of addition of sequences to multiple alignment is important, since it
influences the number of gaps in the alignment.
So once pairwise alignments are produced (including gaps) gap penalties are
reweighted so that gap penalties are less in poorly conserved regions which
already contain gaps)
Gaps are also weighted differently in hydrophilic vs. hydrophobic regions, since
hydrophobic regions tend to be buried secondary structures, but hydrophilic
regions are more exposed and variable.
All of the weighting features tend to help confine gaps to the regions of proteins
where they would be expected, those not in alpha or beta structure.
If the secondary structure of one or more members of the alignment is known,
this information can be used to further weight the alignment to minimize disruption
of secondary structure
Sequences are also weighted so that those that diverged from the phylogenetic
tree near the root have greater weight than those that diverged further out
on the tree.
Progressive programs are only as good as the initial pairwise alignments, so
errors can be propagated throughout the alignment.
Iterative programs recalculate initial pairwise alignments later to improve
pairwise and overall alignments. MultAlin
is an example.
PSI-BLAST
Iterative version of BLAST.
Hits above a threshold E score are used to guide produce position specific scoring
matrix(PSSM) which reflects signature conserved features of the proteins.
This PSSM is used to query a protein database to find additional proteins that
fit the PSSM. New matches above threshold are added to PSSM for next round.
Database is can be requeried until no additional hits above threshold are found.
biggest advantage of PSI-BLAST is not necessarily for the quality of the alignment,
but its sensitivity for identifying essentially all matches in the database.
BLAST itself produces multiple
alignments, but they are based on initial query and thus not necessarily optimal.
Can produce excellent alignments, provided enough sequences are available.
Number of sequences required depends on diversity of sequence set, since more
diverse sequences may make identification of patterns of conservation more difficult.
HMM program (such as HMMer) is first
trained on a set of sequences
The training set need not be aligned, but it can be.
Other input data can be amino acid distribution data or substitution matrices.
Input data are used to identify statistical weights for amino acid identities,
as well as gaps and deletions, for each position in the model.
Training sequences are then aligned to the model and the model readjusted to
provide the best fit to the input sequences.
This step is repeated until no further improvement is made.
Additional sequences can then be fed into the system to align them to the training
sequences. Model can be adjusted during this step as well.
Model can be used to produce Position Specific scoring matrix (PSSM)
PSSMs are log odds scores of finding a particular amino acid at a particular
position in a protein sequence, much as the protein substitution matrices were
log odds scores of substitution of one amino acid for another.
PSSM can then be used to identify new protein sequences which are related to
the known aligned proteins
Need convenient means to manipulate and display multiple sequence alignments.
Most programs have basic text output, with symbols to flag conserved residues
Often convenient to rearrange order of sequences, color code them by residue
type, or box conserved regions.
May also need to do some manual tweaking and editing of alignment.
A number of programs available for different types of computers
You can try looking at the fast clustal alignment or the slow clustal alignment by copying and pasting into one of these viewers.
JalView
is Java-based editor available at the CLUSTALW web site. Since it is Java, it
can be downloaded in your web browser and launched with your CLUSTAL alignment.
Can use to manipulate alignment and make PostScript or GIF files for presentation.
Can also save edited alignment in FASTA format for later use. Jalview can also
be downloaded for stand-alone use.
Other available programs
CINEMA is also Java-based.
you must upload your alignments to a server
Sequence Manipulation Site has easy interface for a variety of sequence ... manipulations
ClustalX is a clustal viewer program with a nice interface, available for most computer platforms.
SeqPup
Initially Mac only, but has been ported to Java for all platforms. Is a stand
alone program (it doesn't require browser.)
BioEdit is a similar
program for PCs only
SeqLogo will
produce graph which displays conserved amino acids of a motif from a PSSM or
alignment.
Boxshade displays
alignments with conserved regions boxed for ease in identification and presentation.