The use of comparison of biological sequence information to infer the relationships
of the existing organisms from which it was derived.
Can be used as the basis for powerful analysis of RNA structure, besides telling about history of organisms.
Relationships are usually represented by "tree" diagram, with branches occurring where a mutation resulted in a different sequence in the progeny form that of the ancestor.
Assume that:
Analysis of non-orthologs can be complicated, especially if you don't realize they aren't orthologs.
One of the best-studied examples of phylogenetic analysis is the rRNA of the small ribosomal subunit (ssu-rRNA). The reasons for this include:
ssu-rRNA has conserved sequences very close to 5' and 3' ends of RNA, so PCR primers can be designed to amplify almost the entire gene from essentially any organism.
So you can analyze the sequences of a known organism, or you can identify what organisms are growing in a population in strange environments:

Yellowstone Park Octopus Spring picture from Jim Brown, NCSU. Click here or on picture for full-sized original.

Close-up of bacterial mats from Octopus Spring also from Jim Brown. The water coming out of the spring is near boiling and cools as it flows down stream. The colored regions are microbial growth.

Obsidian Pool, Yellowstone, from Jim Brown. Note the boiling slurry of water and minerals at the bottom. The analysis was complicated by contamination of samples with runoff from the valley that included microbes from the gut of the bison grazing nearby.
So water, sludge or microbial mat samples were taken and DNA extracted for amplification. You can thus identify rRNA from organisms that you don't know how to cultivate.
Quantitation of differences in aligned sequences relies upon quantitative model
of probability of a particular substitution. Typically, these are the same types
of matrices that are used to create the alignment. Common examples for protein
sequences would be BLOSUM and PAM. For protein coding regions, matrices exist
for all 61 codons. For non-coding nucleic acids, matrices that take into account
the higher frequency of transition (purine -> purine or pyrimidine ->
pyrimidine [R->R or Y->Y]) substitutions than transversion (Y->R or R->Y) mutations.
A similarity matrix is constructed from the alignment scores.
Sequence similarities are converted to differences (1-similarity) since the
differences are indicative of evolutionary distance.
But the differences you observe could be the result of more than one mutation:
e.g. a G in organism 1 and a C in organism 2 could arise by a direct G-> C mutation, or there could have been more steps: G->A->C
also, even unrelated nucleotide sequences are 25% similar by chance
So differences are adjusted upward to estimate evolutionary distance
Tree building proceeds, using the evolutionary distances to construct branches between organisms.
Neighbor joining: building tree by constructing branches between most closely related organisms, then working you way out the distance matrix.
Parsimony: Seeks tree that requires the least number of changes to explain differences
Maximum likelihood: Examines data and looks for model which is most likely to produce it. Looks at differences in variability within alignment and assigns substitution values accordingly.
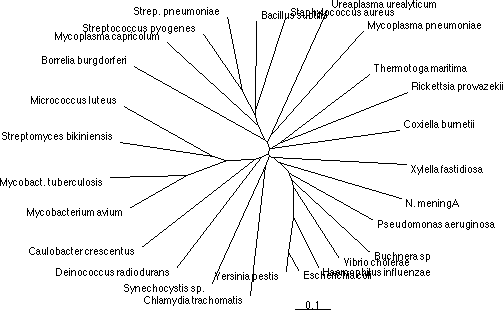
Trees can be arranged as:
 |
 |
|
Dendrograms
|
Phenograms
|
By choosing an outgroup sequence to include with the analysis, you can "root" your tree. Rooting allows you to identify where the sequences analyzed diverged from their common ancestor. The outgroup is a distantly related sequence that is analyzed along with the other sequences, but is known to fall outside the group, such as a eukaryotic ortholog for looking at bacterial sequences.
Looking for key features of sequences from particular groups of organisms. Jim Brown's lecture on RNase P signatures.
Given the possibilities for error in data selection, the final tree must be evaluated for validity. Sometimes this can be as simple as visual inspection. If your tree says the closest relative of a mouse is Salmonella, then maybe you used a mitochondrial sequence instead of a nuclear sequence for the mouse. Or perhaps there was horizontal transfer of a gene. Or you might have ambiguous data.
In some cases a bootstrap analysis is used to look for biases in the data. The analysis is rerun numerous times using random subsets of the data. Parts of the tree that occur nearly every time are reliable, ones that occur only occasionally are suspect.
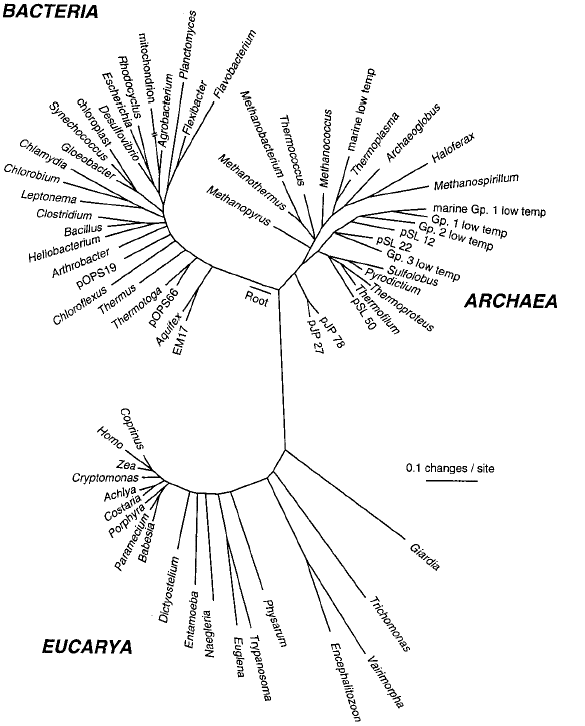
The Big Tree of Life from Norm Pace. From: The universal nature of biochemistry. Proc Natl Acad Sci U S A. 2001 98:805-808. Also see his microbial diversity pages
Phylogenetic Comparative Analysis of RNA structure:
Sequence homologous RNAs from a variety of organisms
Align sequences
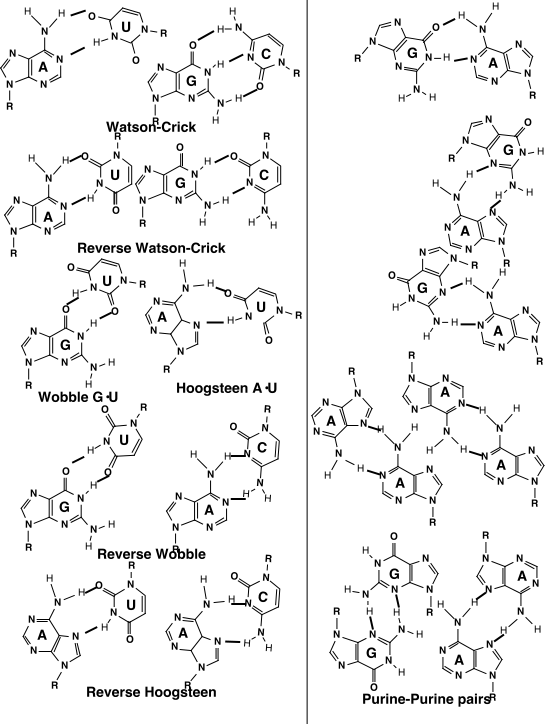
Look for covariation of base sequences as evidence of interaction
(i.e. W-C or other pairs)
|-|
U<-- A-U -->A
|-|
A<-- C-G -->U
|-|
Complementarity maintained by compensatory base changes.
Can also detect some non Watson-Crick tertiary interactions this way.
(Computer programs aid in aligning seqs. and detecting covariations)
Also identifies highly conserved sequences that may be required for special
functions.
BUT: Cannot tell base-pairing of conserved residues that don't
vary
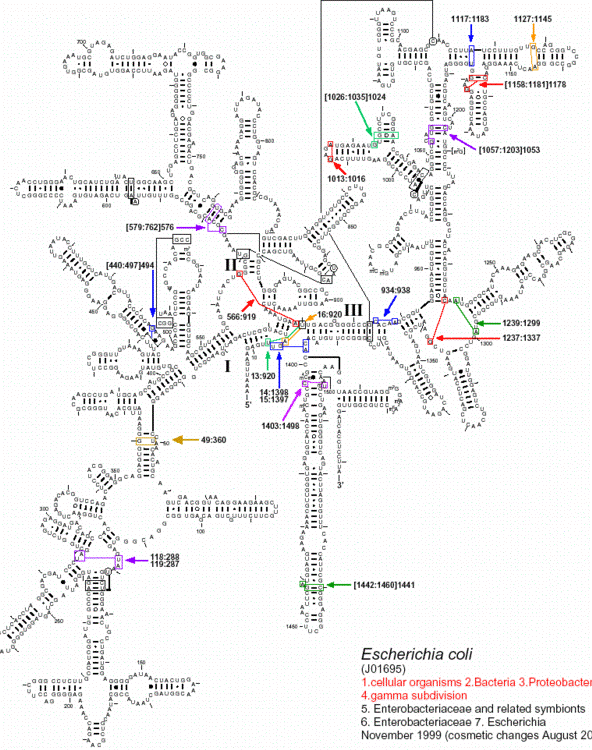
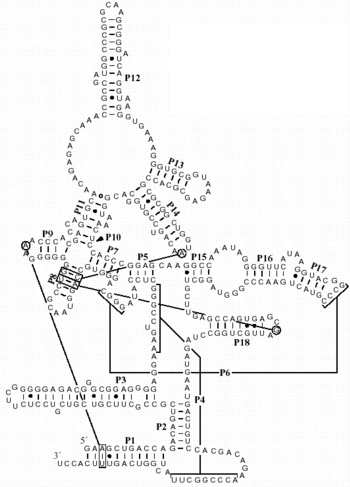
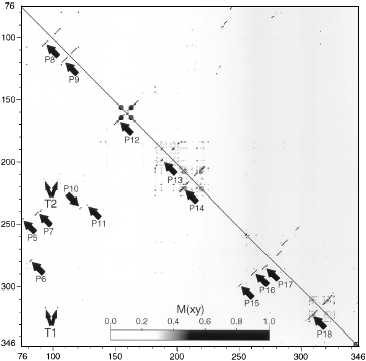
Examples of well-defined secondary structures derived from phylogenetic
analysis:
tRNA
rRNA (16S, 23S, 5S)
Group I intron
Group II intron
RNase P
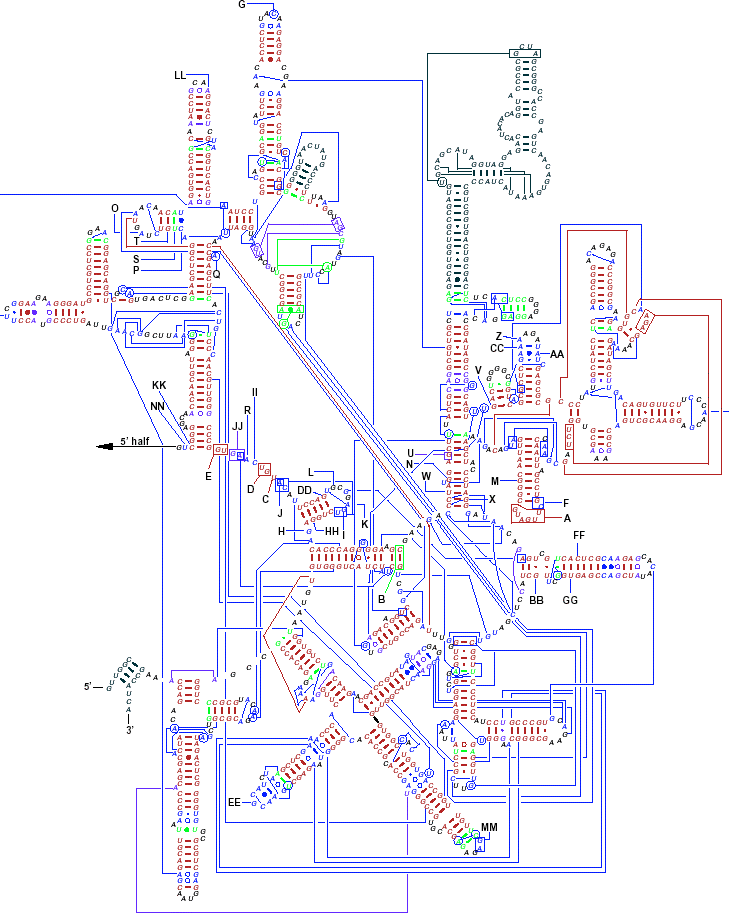
Pairings shown in red were predicted by phylogenetics and observed in the crystal
structure.
Green pairs were predicted, but not observed.
Blue pairs were observed, but not predicted.
 |
|
Computer prediction based on sequence
Not as straightforward as you would think
Look for regions of molecule that can form W-C pairs with a given region.
But one region of RNA may be complementary to many other short stretches
of sequence.
Computer algorithms can help see which folding choices yield lowest
free energy. (Zuker, 1989)
e.g. G-C pair more favorable than A-U
G•U (wobble) pair is almost as favorable as A-U
(makes for even more regions of complementarity
Also takes into account "nearest neighbor" rules for stacking:
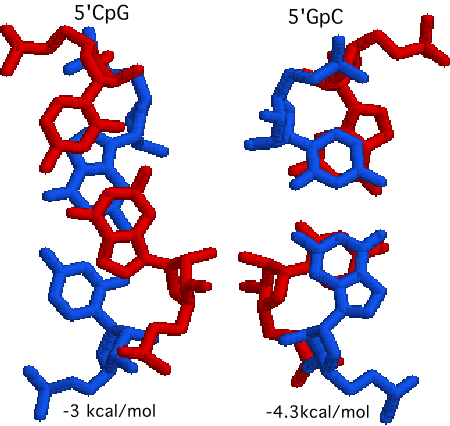
So identical nucleotide compositions can have different energies due
to order of sequence.
Unpaired structures are unfavorable (loops, bulges).
But some are more unfavorable than others.
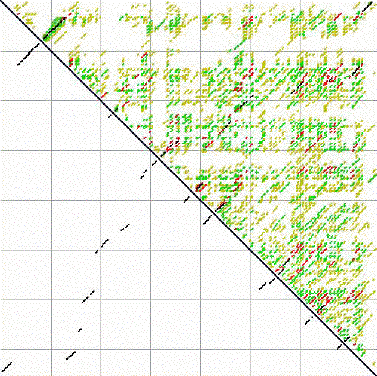
Even with all of this info, still not always very accurate:
Many regions of molecule not involved in W-C pairs, form "non-canonical"
pairs instead: e.g. G-A, A-C, etc.
Non-canonical pairs can have very favorable energy, but many don't tend
to fit A-form helix very well.
Phylogenetics can sometimes predict non-canonicals
Many predicted non-canonical pairs have been confirmed by crystal structures.
Also 2'OH is often involved in tertiary pairs, but it is difficult to
predict, since it is both an H-bond donor and acceptor.
|
|
|
|
|
Self-complementarity |
Computer prediction |
Phylogenetic |