

Alignment of DNA or protein sequences in bioinformatics serves as a number of
purposes:
DNA sequence data can be aligned to assemble larger contiguous DNA sequences
Protein sequences can be aligned to identify important regions, such as active sites
DNA sequences can be aligned to identify regulatory control regions
Alignment of any two sequences implies that those sequences are related.
Overlapping DNA sequence reads are derived from a single DNA molecule
Protein, RNA, or DNA sequences from a particular gene for different organisms are related by the ancestry of those organisms (orthologs)
Duplicated genes (and pseudogenes) within an organism are related to each other or a particular parental gene (paralogs)
Molecular sequences that are not related in some way, do not produce statistically
significant sequence alignments
A key then, to answering many bioinformatics questions lies in the ability to reliably align sequence information
Sequences can be aligned:
Globally, in which as many characters as possible over the entire sequence are aligned
Or locally, in which regions of sequences which are highly similar are aligned
Sequences can be compared in pairs or as multiple alignments of related sequences
In practice, these alignments are performed similarly, and can be understood
in the context of a dot-matrix sequence comparison.
Dot matrix analysis displays the primary sequence of pairs of proteins on the X and Y axes of a graph.
Dots are plotted on the graph where the X and Y coordinate sequences are identical.
Regions of identical sequence are revealed as diagonal rows of dots.
Random matches are seen as isolated dots.
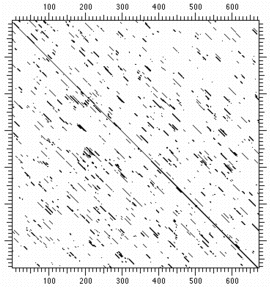
A comparison of DNAs sequences for elongation factor EF-G from
S. aureus and T.
thermophilus is shown below left. A dot is placed every time a nucleotide
in the S. aureus sequence matches that of T. thermophilus. Since there are only
four types of nucleotides, random matches occur many places throughout the sequence
and obscure regions of significant similarity.
A similar analysis of the corresponding peptide sequences is shown at right.
Random matches of the 20 amino acids also occur, but diagonal regions of sequence
similarity are all more discernible.
|
|
| DNA alignment - single nucleotide identity | Protein alignment - single residue identity |
*- So if you need to align protein coding regions of DNA, translate them first into protein, for most sensitive and accurate alignment.
Random matches can be filtered by using a sliding window to compare sequences. In this case, a block of characters is compared between sequences. A dot is placed on the alignment only if a threshold value of matches is detected.
An example is shown below on a small portion of the amino acid sequence. At left, a one or dot is placed wherever the sequences match.
In the center panel, a score is placed for the number of matches between sequences for each block of 3 amino acids.
For 3 consecutive matches, the score is 3
For 2 out of 3 the score is 2
For 1 out of three the score is 1
While there are more 1's on the second matrix, by ignoring all values below 2 (right panel), the random matches are ignored.
|
|
|
||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|
|
Word Size = 3
|
|
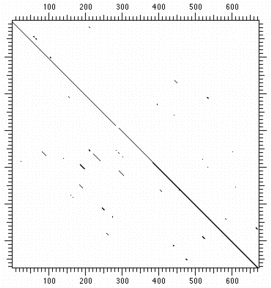
The full matrix for the proteins is shown below:
|
 |
| Protein- single residue identity | Protein identity 5 residues of 23 |
Even using a low stringency window greatly improves the signal/nose ratio. One can readily see a diagonal stretching across the matrix indicating sequence similarity throughout the length of both sequences. This works well for DNA sequences, but you must use higher stringencies; longer lengths help too:
|
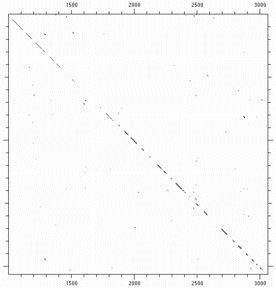 |
| DNA alignment - single nucleotide identity | DNA alignment - 15 of 23 identical |
Going back to proteins:
You can also compare a sequence with itself to look for repeated sequences:

Protein sequence of porcine submaxillary mucin compared to itself looking for exact matches of 39 residues.
You can see diagonal of unique sequence at termini.
In between are 135.6 repeats of an 81 residue sequence, extending 10991 amino acids, this is an extreme case.
Repeats are often detectable in comparing related sequences to each other.
Dot matrix analysis makes them obvious.
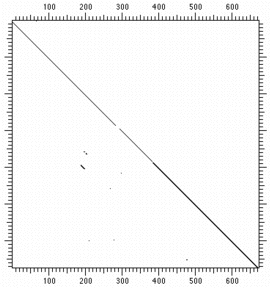
The alignment can be improved by raising the score for a match to +6 and penalizing
a mismatch with -1:
|
 |
| Protein identity 5 residues of 23(match=1, mismatch=0) | Protein identity 5 residues of 23(match=6, mismatch=-1) |
Virtually all the background matches have been eliminated. Even better results can be obtained using weighted scoring matrices.
Comparison of related proteins revealed that substitution of chemically similar amino acid residues was fairly common in many positions of the protein sequences.
Other substitutions were found to be less common
Some were extremely rare.
Substitution matrices quantitate these differences in substitution frequencies for use in evaluation of alignments
|
 |
|
Protein identity 5 residues of 23
(match=6, mismatch=-1) |
Protein BLOSUM62 matrix
5 residues of 23 match |
Where do scoring matrices come from?
Two common types of matrix: PAM and BLOSUM
Derived from manual alignment of 71 groups of similar protein sequences
Each group >85% similar within group
All members of group similar in function
so mutations present didn't affect protein function => "Accepted"
Mutations
Organized sequences based on presumed phylogeny of sequences
then identified changes from one amino acid to another by mutation
Mutation probability = # amino acid changes / exposure to mutation (exposure = amino acid frequency/total #aa changes/100 sites)
summed for all groups
NSDE - most mutable amino acids
CW - least mutable amino acids (unique chemistry)PAM1 = 1 percent accepted mutation
Other versions extrapolated from PAM1
PAM250 =250% change in sequence overall (~2500 million years) -> ~20% similarity
PAM120= 40% similarity
PAM80 = 50% similarity
PAM60 = 60% similarity
Choosing PAM matrix appropriate for similarity gives more reliable score for alignments
The PAM250 Matrix
| C | S | T | P | A | G | N | D | E | Q | H | R | K | M | I | L | V | F | Y | W | |
| C | 12 | |||||||||||||||||||
| S | 0 | 2 | ||||||||||||||||||
| T | -2 | 1 | 3 | |||||||||||||||||
| P | -3 | 1 | 0 | 6 | ||||||||||||||||
| A | -2 | 1 | 1 | 1 | 2 | |||||||||||||||
| G | -3 | 1 | 0 | 0 | 1 | 5 | ||||||||||||||
| N | -4 | 1 | 0 | 0 | 0 | 0 | 2 | |||||||||||||
| D | -5 | 0 | 0 | -1 | 0 | 1 | 2 | 4 | ||||||||||||
| E | -5 | 0 | 0 | -1 | 0 | 0 | 1 | 3 | 4 | |||||||||||
| Q | -5 | -1 | -1 | 0 | 0 | -1 | 1 | 2 | 2 | 4 | ||||||||||
| H | -3 | -1 | -1 | 0 | -1 | -2 | 2 | 1 | 1 | 3 | 6 | |||||||||
| R | -4 | 0 | -1 | 0 | -2 | -3 | 0 | -1 | -1 | 1 | 2 | 6 | ||||||||
| K | -5 | 0 | 0 | -1 | -1 | -2 | 1 | 0 | 0 | 1 | 0 | 3 | 5 | |||||||
| M | -5 | -2 | -1 | -2 | -1 | -3 | -2 | -3 | -2 | -1 | -2 | 0 | 0 | 6 | ||||||
| I | -2 | -1 | 0 | -2 | -1 | -3 | -2 | -2 | -2 | -2 | -2 | -2 | -2 | 2 | 5 | |||||
| L | -6 | -3 | -2 | -3 | -2 | -4 | -3 | -4 | -3 | -2 | -2 | -3 | -3 | 4 | 2 | 6 | ||||
| V | -2 | -1 | 0 | -1 | 0 | -1 | -2 | -2 | -2 | -2 | -2 | -2 | -2 | 2 | 4 | 2 | 4 | |||
| F | -4 | -3 | -3 | -5 | -3 | -5 | -3 | -6 | -5 | -5 | -2 | -4 | -5 | 0 | 1 | 2 | -1 | 9 | ||
| Y | 0 | -3 | -3 | -5 | -3 | -5 | -2 | -4 | -4 | -4 | 0 | -4 | -4 | -2 | -1 | -1 | -2 | 7 | 10 | |
| W | -8 | -2 | -5 | -6 | -6 | -7 | -4 | -7 | -7 | -5 | -3 | 2 | -3 | -4 | -5 | -2 | -6 | 0 | 0 | 17 |
What the scores mean:
Score=0: odds of substitution of one amino acid with the other equals that of random chance
Score>0: substitutions occur more likely than by chance (i.e. evolutionarily selected for)
Score<0: substitutions occur less likely than by chance (i.e. evolutionarily selected against)
Where the numbers come from:
For example : F <-> Y substitutions
In PAM1 mutation frequencies:
| mutation |
PAM1
|
PAM250
|
| F -> Y |
0.0021
|
.015
|
| F -> F |
0.9946
|
0.32
|
| Y -> F |
0.0028
|
0.20
|
| Y -> Y |
0.9946
|
0.31
|
so for PAM250
F->Y probability (0.15) divided by frequency of F in data set (0.040) = relative
frequency of change (3.75)
log(3.75)= 0.57 X10 = 5.7
Y->F probability (0.20) X freq. Y (0.030) = 6.7
log(6.7)= 0.83 X10 =8.3
Average of F->Y and Y->F= (5.7+8.3)/2= 7 (the value in the table above)
Table values are log odds scores, so odds of adding successive characters to alignment is sum of log probability (product of probabilities) of each independent event.
Since PAM is extrapolated from closely related sequences, may not be best way to look at more distant relatives. BLOSUM matrix is based upon the BLOCKS database of protein motifs
e.g. RNase P BLOCK AxxRNR...
what are the frequencies of substitutions at the xx positions?
Allows alignment of short regions of sequence from very divergent proteins
2000 blocks of conserved patterns from 500 different families of proteins
Non-conserved amino acids counted and frequency of substitution pairs counted
Closely related proteins can cause over representation of sequences, so these
sequences were merged into a single consensus
BLOSUM62: 62% of residues in data set identical
BLOSUM62 Matrix
| C | S | T | P | A | G | N | D | E | Q | H | R | K | M | I | L | V | F | Y | W | |
| C | 9 | |||||||||||||||||||
| S | -1 | 4 | ||||||||||||||||||
| T | -1 | 1 | 5 | |||||||||||||||||
| P | -3 | -1 | -1 | 7 | ||||||||||||||||
| A | 0 | 1 | 0 | -1 | 4 | |||||||||||||||
| G | -3 | 0 | -2 | -2 | 0 | 6 | ||||||||||||||
| N | -3 | 1 | 0 | -2 | -2 | 0 | 6 | |||||||||||||
| D | -3 | 0 | -1 | -1 | -2 | -1 | 1 | 6 | ||||||||||||
| E | -4 | 0 | -1 | -1 | -1 | -2 | 0 | 2 | 5 | |||||||||||
| Q | -3 | 0 | -1 | -1 | -1 | -2 | 0 | 0 | 2 | 5 | ||||||||||
| H | -3 | -1 | -2 | -2 | -2 | -2 | 1 | -1 | 0 | 0 | 8 | |||||||||
| R | -3 | -1 | -1 | -2 | -1 | -2 | 0 | -2 | 0 | 1 | 0 | 5 | ||||||||
| K | -3 | 0 | -1 | -1 | -1 | -2 | 0 | -1 | 1 | 1 | -1 | 2 | 5 | |||||||
| M | -1 | -1 | -1 | -2 | -1 | -3 | -2 | -3 | -2 | 0 | -2 | -1 | -1 | 5 | ||||||
| I | -1 | -2 | -1 | -3 | -1 | -4 | -3 | -3 | -3 | -3 | -3 | -3 | -3 | 1 | 4 | |||||
| L | -1 | -2 | -1 | -3 | -1 | -4 | -3 | -4 | -3 | -2 | -3 | -2 | -2 | 2 | 2 | 4 | ||||
| V | -1 | -2 | 0 | -2 | 0 | -3 | -3 | -3 | -2 | -2 | -3 | -3 | -2 | 1 | 3 | 1 | 4 | |||
| F | -2 | -2 | -2 | -4 | -2 | -3 | -3 | -3 | -3 | -3 | -1 | -3 | -3 | 0 | 0 | 0 | -1 | 6 | ||
| Y | -2 | -2 | -2 | -3 | -2 | -3 | -2 | -3 | -2 | -1 | 2 | -2 | -2 | -1 | -1 | -1 | -1 | 3 | 7 | |
| W | -2 | -3 | -2 | -4 | -3 | -2 | -4 | -4 | -3 | -2 | -2 | -3 | -3 | -1 | -3 | -2 | -3 | 1 | 2 | 11 |
Difference between PAM250 and BLOSUM62
| C | S | T | P | A | G | N | D | E | Q | H | R | K | M | I | L | V | F | Y | W | |
| C | 3 | |||||||||||||||||||
| S | 1 | -2 | ||||||||||||||||||
| T | -1 | -2 | ||||||||||||||||||
| P | 2 | 1 | -1 | |||||||||||||||||
| A | -2 | 1 | 2 | -2 | ||||||||||||||||
| G | 1 | 2 | 2 | 1 | -1 | |||||||||||||||
| N | -1 | 2 | 2 | -4 | ||||||||||||||||
| D | -2 | 1 | 2 | 2 | 1 | -2 | ||||||||||||||
| E | -1 | 1 | 1 | 2 | 1 | 1 | -1 | |||||||||||||
| Q | -2 | -1 | 1 | 1 | 1 | 1 | 2 | -1 | ||||||||||||
| H | 1 | 2 | 1 | 1 | 2 | 1 | 3 | -2 | ||||||||||||
| R | -1 | 1 | 2 | -1 | -1 | 1 | -1 | 2 | 1 | |||||||||||
| K | -2 | 1 | 1 | 1 | -1 | 1 | 1 | |||||||||||||
| M | -4 | -1 | -1 | 1 | 1 | 1 | ||||||||||||||
| I | -1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | ||||||
| L | -5 | -1 | -1 | -1 | 1 | -1 | -1 | 2 | 2 | |||||||||||
| V | -1 | 1 | 1 | 2 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | |||||||||
| F | -2 | -1 | -1 | -1 | -1 | -2 | -3 | -2 | -2 | -1 | -1 | -2 | 1 | 2 | 3 | |||||
| Y | 2 | -1 | -1 | -2 | -1 | -2 | -1 | -2 | -3 | -2 | -2 | -2 | -1 | -1 | 4 | 3 | ||||
| W | -6 | 1 | -3 | -2 | -3 | -5 | -3 | -4 | -3 | -1 | 5 | -3 | -2 | -3 | -1 | -2 | 6 |
So different scoring matrix can have an effect on how the alignment result
Can also make PAM scoring matrix for nucleic acids
At 1PAM: Mutation frequency matrix
| A | G | T | C | |
| A | 0.99 | |||
| G | 0.0033 | 0.99 | ||
| T | 0.0033 | 0.0033 | 0.99 | |
| C | 0.0033 | 0.0033 | 0.0033 | 0.99 |
so log odds scoring matrix:
| A | G | T | C | |
| A | 2 | |||
| G | -6 | 2 | ||
| T | -6 | -6 | 2 | |
| C | -6 | -6 | -6 | 2 |
These values can be weighted to account for higher rates of transition mutations than transversions.
Gap costs: Introducing gaps can aid in aligning sequences, but if you
have unlimited gaps, you can align any 2 sequences. Also, gaps in a protein
structure mean insertion or deletion of structural elements. Can be very deleterious
to protein structure compared to amino acid substitution. So there are penalties
for making gaps, and a separate penalty for extending the gap.
e.g.
AGGTTCGACT
--GT-C-A-T
Usually penalty is high for opening a new gap, but is smaller for extending a gap. (If a gap is actually allowed in a protein sequence, such as variation in the size of a loop, the size difference is probably not critical). Gap and extension penalties vary with matrix used. Usually default values for a given scoring matrix work best.
Most commonly used algorithms are FASTA and BLAST
Both are useful for searching for matches of your sequence to a large database, such as Genbank
To do so, both use simplifying shortcuts to minimize computational time without sacrificing much sensitivity
Sequences are broken into words (also called k-tuples)
Matches between word characters are detected, offset of positions calculated
Series of character matches with same offset = a hit
Additional matches on same diagonal are identified -> diagonal matches
Diagonal matches within certain distance of other matches are joined using gaps
Joined regions are evaluated using substitution matrix scores
FASTA alignment of EF-G sequences using BLOSUM50 matrix (for very divergent
sequences)
>>gi|119190|sp|P13551|EFG_THETH ELONGATION FACTOR G (EF-G) (691 aa)
initn: 2949 init1: 1359 opt: 2943 Z-score: 3250.5 bits: 611.9 E(): 3.1e-174
Smith-Waterman score: 2943; 62.591% identity (63.050% ungapped) in 687 aa overlap (4-688:6-689)
Entrez lookup Re-search database General re-search
>gi|119 4- 688:---------------------------------------------------------------------:
10 20 30 40 50 60 70
gi|692 MAREFSLEKTRNIGIMAHIDAGKTTTTERILYYTGRIHKIGETHEGASQMDWMEQEQDRGITITSAATTAAWEGHRVN
:..:.. ::::: ::::::::::::::::::::::::::.::::. ::.::::..::::::.:.:: :. ::.:
gi|119 MAVKVEYDLKRLRNIGIAAHIDAGKTTTTERILYYTGRIHKIGEVHEGAATMDFMEQERERGITITAAVTTCFWKDHRIN
10 20 30 40 50 60 70 80
80 90 100 110 120 130 140 150
gi|692 IIDTPGHVDFTVEVERSLRVLDGAVTVLDAQSGVEPQTETVWRQATTYGVPRIVFVNKMDKLGANFEYSVSTLHDRLQAN
:::::::::::.:::::.::::::..:.:...:::::.::::::: : ::::.:.::::: ::.. . :...:: :
gi|119 IIDTPGHVDFTIEVERSMRVLDGAIVVFDSSQGVEPQSETVWRQAEKYKVPRIAFANKMDKTGADLWLVIRTMQERLGAR
90 100 110 120 130 140 150 160
160 170 180 190 200 210 220 230
gi|692 AAPIQLPIGAEDEFEAIIDLVEMKCFKYTNDLGTEIEEIEIPEDHLDRAEEARASLIEAVAETSDELMEKYLGDEEISVS
. .::::: :: : .:::...:: . : :::::.:.:: :::..::.:.: . .:.:..:. ....: ::: :: .
gi|119 PVVMQLPIGREDTFSGIIDVLRMKAYTYGNDLGTDIREIPIPEEYLDQAREYHEKLVEVAADFDENIMLKYLEGEEPTEE
170 180 190 200 210 220 230 240
240 250 260 270 280 290 300 310
gi|692 ELKEAIRQATTNVEFYPVLCGTAFKNKGVQLMLDAVIDYLPSPLDVKPIIGHRASNPEEEVIA-KADDSAEFAALAFKVM
:: :::..: .... ::. :.:.:::::::.::::.::::::::. :: : ..:: ::. . : .. .::::::.:
gi|119 ELVAAIRKGTIDLKITPVFLGSALKNKGVQLLLDAVVDYLPSPLDIPPIKG---TTPEGEVVEIHPDPNGPLAALAFKIM
250 260 270 280 290 300 310
320 330 340 350 360 370 380 390
gi|692 TDPYVGKLTFFRVYSGTMTSGSYVKNSTKGKRERVGRLLQMHANSRQEIDTVYSGDIAAAVGLKDTGTGDTLCGEKND-I
.:::::.:::.::::::.:::::: :.:::..:::.:::.:::: :.:.. . .::..:.::::.: ::::: :: .
gi|119 ADPYVGRLTFIRVYSGTLTSGSYVYNTTKGRKERVARLLRMHANHREEVEELKAGDLGAVVGLKETITGDTLVGEDAPRV
320 330 340 350 360 370 380 390
400 410 420 430 440 450 460 470
gi|692 ILESMEFPEPVIHLSVEPKSKADQDKMTQALVKLQEEDPTFHAHTDEETGQVIIGGMGELHLDILVDRMKKEFNVECNVG
::::.: ::::: ...:::.::::.:..:::..: ::::::.. : ::::.::.:::::::.:.:::.:.::.:. :::
gi|119 ILESIEVPEPVIDVAIEPKTKADQEKLSQALARLAEEDPTFRVSTHPETGQTIISGMGELHLEIIVDRLKREFKVDANVG
400 410 420 430 440 450 460 470
480 490 500 510 520 530 540 550
gi|692 APMVSYRETFKSSAQVQGKFSRQSGGRGQYGDVHIEFTPNETGAGFEFENAIVGGVVPREYIPSVEAGLKDAMENGVLAG
:.:.::::. . ..:.::: ::.::::::: :.:. : :.:::: :::::::.:.::::.:. :...::..: : :
gi|119 KPQVAYRETITKPVDVEGKFIRQTGGRGQYGHVKIKVEPLPRGSGFEFVNAIVGGVIPKEYIPAVQKGIEEAMQSGPLIG
480 490 500 510 520 530 540 550
560 570 580 590 600 610 620 630
gi|692 YPLIDVKAKLYDGSYHDVDSSEMAFKIAASLALKEAAKKCDPVILEPMMKVTIEMPEEYMGDIMGDVTSRRGRVDGMEPR
.:..:.:. :::::::.:::::::::::.:.:.:::..: :::::::.:.: . :::::::..::...:::.. :::::
gi|119 FPVVDIKVTLYDGSYHEVDSSEMAFKIAGSMAIKEAVQKGDPVILEPIMRVEVTTPEEYMGDVIGDLNARRGQILGMEPR
560 570 580 590 600 610 620 630
640 650 660 670 680 690
gi|692 GNAQVVNAYVPLSEMFGYATSLRSNTQGRGTYTMYFDHYAEVPKSIAEDIIKKNKGE
:::::. :.:::.:::::::.:::.:::::...:.:::: ::::.. : .::
gi|119 GNAQVIRAFVPLAEMFGYATDLRSKTQGRGSFVMFFDHYQEVPKQVQEKLIKGQ
640 650 660 670 680 690
Different matrix can give different alignment and score. Compare BLOSUM50
alignment:
Gap penalty: -10 extension: -2
280 290 300 310
LPSPLDVKPIIGHRASNPEEEVIA-KADDSAEFAALAFKVM
::::::. :: : ..:: ::. . : .. .::::::.:
LPSPLDIPPIKG---TTPEGEVVEIHPDPNGPLAALAFKIM
With PAM120 alignment (less divergent)
Gap penalty: -16 extension: -4
LPSPLDVKPIIGHRASNPEEEVIAKADDSAEFAALAFKVMT
::::::. :: : .. : : .: .. .::::::.:.
LPSPLDIPPIKG--TTPEGEVVEIHPDPNGPLAALAFKIMA
290 300 310
Variations of FASTA program are available for protein-protein, DNA-DNA comparisons. Also DNA-protein comparison by translating DNA.
Works similarly to FASTA, with some differences
First, regions of sequence that are low complexity (like ST-rich regions, which
are easily substituted) are filtered out
Then a list of overlapping 3 or 4 letter words are derived from sequence
Substitution score is used to identify high scoring words above a threshold
value
-> small number of words (~50) to compare to database
Database already indexed for high-scoring words
Matches within given distance of other matches on same diagonal are used to
align sequences
BLAST alignment of S. aureus (Query) vs T. thermophilus (Subject)
Query: 4 EFSLEKTRNIGIMAHIDAGKTTTTERILYYTGRIHKIGETHEGASQMDWMEQEQDRGXXX 63
E+ L++ RNIGI AHIDAGKTTTTERILYYTGRIHKIGE HEGA+ MD+MEQE++RG
Sbjct: 6 EYDLKRLRNIGIAAHIDAGKTTTTERILYYTGRIHKIGEVHEGAATMDFMEQERERGITI 65
np-binding 19 ********
Query: 64 XXXXXXXXWEGHRVNIIDTPGHVDFTVEVERSLRVLDGAVTVLDAQSGVEPQTETVWRQA 123
W+ HR+NIIDTPGHVDFT+EVERS+RVLDGA+ V+D+ GVEPQ+ETVWRQA
Sbjct: 66 TAAVTTCFWKDHRINIIDTPGHVDFTIEVERSMRVLDGAIVVFDSSQGVEPQSETVWRQA 125
np-binding 83 *****
Query: 124 TTYGVPRIVFVNKMDKLGANFEYSVSTLHDRLQANAAPIQLPIGAEDEFEAIIDLVEMKC 183
Y VPRI F NKMDK GA++ + T+++RL A +QLPIG ED F IID++ MK
Sbjct: 126 EKYKVPRIAFANKMDKTGADLWLVIRTMQERLGARPVVMQLPIGREDTFSGIIDVLRMKA 185
np-binding 137 ****
Query: 184 FKYTNDLGTEIEEIEIPEDHLDRAEEARASLIEAVAETSDELMEKYLGDEEISVSELKEA 243
+ Y NDLGT+I EI IPE++LD+A E L+E A+ + +M KYL EE + EL A
Sbjct: 186 YTYGNDLGTDIREIPIPEEYLDQAREYHEKLVEVAADFDENIMLKYLEGEEPTEEELVAA 245
Query: 244 IRQATTNVEFYPVLCGTAFKNKGVQLMLDAVIDYLPSPLDVKPIIGHRASNPEEEVIA-K 302
IR+ T +++ PV+ G+A+KNKGVQL+LDAV+DYLPSPLD+ PI G + PE EV+
Sbjct: 246 IRKGTIDLKITPVFLGSALKNKGVQLLLDAVVDYLPSPLDIPPIKG---TTPEGEVVEIH 302
Query: 303 ADDSAEFAALAFKVMTDPYVGKLTFFRVYSGTMTSGSYVKNSTKGKRERVGRLLQMHANS 362
D + +AALAFK+M DPYVG+LTF RVYSGT+TSGSYV N+TKG++ERV RLL+MHAN
Sbjct: 303 PDPNGPLAALAFKIMADPYVGRLTFIRVYSGTLTSGSYVYNTTKGRKERVARLLRMHANH 362
Query: 363 RQEIDTVYSGDIAAAVGLKDTGTGDTLCGEKND-IILESMEFPEPVIHLSVEPKSKADQD 421
R+E++ + +GD+ A VGLK+T TGDTL GE +ILES+E PEPVI +++EPK+KADQ+
Sbjct: 363 REEVEELKAGDLGAVVGLKETITGDTLVGEDAPRVILESIEVPEPVIDVAIEPKTKADQE 422
Query: 422 KMTQALVKLQEEDPTFHAHTDEETGQVIIGGMGELHLDILVDRMKKEFNVECNVGAPMVS 481
K++QAL +L EEDPTF T ETGQ II GMGELHL+I+VDR+K+EF V+ NVG P V+
Sbjct: 423 KLSQALARLAEEDPTFRVSTHPETGQTIISGMGELHLEIIVDRLKREFKVDANVGKPQVA 482
Query: 482 YRETFKSSAQVQGKFSRQSGGRGQYGDVHIEFTPNETGAGFEFENAIVGGVVPREYIPSV 541
YRET V+GKF RQ+GGRGQYG V I+ P G+GFEF NAIVGGV+P+EYIP+V
Sbjct: 483 YRETITKPVDVEGKFIRQTGGRGQYGHVKIKVEPLPRGSGFEFVNAIVGGVIPKEYIPAV 542
Query: 542 EAGLKDAMENGVLAGYPLIDVKAKLYDGSYHDVDSSEMAFXXXXXXXXXXXXXXCDPVIL 601
+ G+++AM++G L G+P++D+K LYDGSYH+VDSSEMAF DPVIL
Sbjct: 543 QKGIEEAMQSGPLIGFPVVDIKVTLYDGSYHEVDSSEMAFKIAGSMAIKEAVQKGDPVIL 602
Query: 602 EPMMKVTIEMPEEYMGDIMGDVTSRRGRVDGMEPRGNAQVVNAYVPLSEMFGYATSLRSN 661
EP+M+V + PEEYMGD++GD+ +RRG++ GMEPRGNAQV+ A+VPL+EMFGYAT LRS
Sbjct: 603 EPIMRVEVTTPEEYMGDVIGDLNARRGQILGMEPRGNAQVIRAFVPLAEMFGYATDLRSK 662
Query: 662 TQGRGTYTMYFDHYAEVPKSIAEDIIK 688
TQGRG++ M+FDHY EVPK + E +IK
Sbjct: 663 TQGRGSFVMFFDHYQEVPKQVQEKLIK 689
Results similar to FASTA using same matrix. XXXXX indicates regions masked for low complexity.
Selecting the BLAST Program
You need to pick the right version of BLAST to suit your data:
| Program | Description |
| blastp | Compares a protein sequence against a protein sequence database. |
| blastn | Compares a nucleotide query sequence against a nucleotide sequence database. |
| blastx | Translates your nucleotide query sequence in all reading frames against and compares these translations against a protein sequence database. You could use this option to find potential translation products of an unknown nucleotide sequence. |
| tblastn | Compares a protein query sequence against a nucleotide sequence database which has been translated in all reading frames. |
| tblastx | Compares the six-frame translations of a nucleotide query sequence against the six-frame translations of a nucleotide sequence database. |
Selecting the BLAST Database (from the BLAST
tutorial)
| Database | Description |
| nr | All non-redundant GenBank CDS translations (open reading frames of nucleotide seq)+PDB+SwissProt+PIR+PRF |
| month | similar to nr for sequences released in the last 30 days. |
| swissprot | The SWISS-PROT protein sequence database. | patents | Protein sequences from the GenBank Patent database. | yeast | Protein translations of the Yeast genome. |
| E. coli | Protein translations of the Escherichia coli genome. |
| pdb | Sequences from the 3-dimensional structure Protein Data Bank. |
| kabat | Kabat's Database of Sequences of Immunological interest. (e.g. IgG's) |
| alu | Translations of Alu repeats from REPBASE |
| Database | Description |
| nr | All non-redundant GenBank+EMBL+DDBJ+PDB sequences (excluding EST, STS, GSS, and HTGS sequences). |
| month | GenBank+EMBL+DDBJ+PDB sequences released in the last 30 days. |
| dbest |
Non-redundant database of GenBank+EMBL+DDBJ ESTs (Expressed sequence tags: sequences of random cDNAs obtained from specific tissue sources.) |
| dbsts |
Non-redundant database of GenBank+EMBL+DDBJ STSs (Sequence Tagged Site: random physical genomic map marker) |
| mouse ests | The non-redundant Database of mouse GenBank+EMBL+DDBJ ESTs. |
| human ests | The non-redundant Database of human GenBank+EMBL+DDBJ ESTs. |
| other ests | The non-redundant database of GenBank+EMBL+DDBJ ESTs from all organisms except mouse and human. |
| yeast | Sequence fragments from the Yeast complete genome. |
| E. coli | Escherichia coli genomic nucleotide sequences. |
| pdb | Sequences from the 3-dimensional structure Protein Data Bank. |
| kabat [kabatnuc] | Kabat's Database of Sequences of Immunological interest. (e.g. IgG's) |
| patents | Protein sequences from the GenBank Patent database. |
| vector | Vector subset of GenBank(R), NCBI, (ftp://ncbi.nlm.nih.gov/pub/blast/db/ directory). |
| mito | Database of mitochondrial sequences (Rel. 1.0, July 1995). |
| alu | Translations of Alu repeats from REPBASE |
| gss | Genome Survey Sequences. |
| htgs | High Throughput Genomic Sequences. |
The Raw Score (S) is the sum of all the matches for a given alignment, minus mismatch and gap penalties. This value tends to be larger for larger proteins, so how can you tell if it is significant?
Expected (E) value is the number of scores that would be expected to exceed the actual score S by chance.
.gif)
where m and n are sequence lengths and K and lambda are scaling factors dependent upon the size of the search and the scoring system.
Probability
(P) of finding a higher score:
P= 1-e(-E)
When E< 0.01, P~E
So the E value is a good measure of the significance of any alignment. Numbers approacing 1 are not likely significant.
Numbers much less than 1 are highly significant.
Smith-Waterman and Needleman-Wunsch use dynamic programming algorithm to find optimal local and global alignments, respectively
Dynamic programming
Identifies highest scoring regions of alignment and extends them to end of alignment.
Highest score for a given reqion nust be made from optimal subscore from step before.
Steps are then traced back from highest score at end of alignment to identify optimal path to that score.
Computationally intensive. Not typically used against databases.
But gives best alignment of two related sequences.
Searches for optimal positioning of gaps to align stretches of similar sequence.
|
|
||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Alignment matrix | Summed using dynamic programming |
# Aligned_sequences: 2
# 1: EFG_THETH
# 2: EFG_STAAM
# Matrix: EBLOSUM62
# Gap_penalty: 10.0
# Extend_penalty: 0.5
#
# Length: 697
# Identity: 430/697 (61.7%)
# Similarity: 542/697 (77.8%)
# Gaps: 10/697 ( 1.4%)
# Score: 2290.0
#
#
#=======================================
EFG_THETH 1 MAVKVEYDLKRLRNIGIAAHIDAGKTTTTERILYYTGRIHKIGEVHEGAA 50
:..|:.|::.|||||.||||||||||||||||||||||||||.||||:
EFG_STAAM 1 MAREFSLEKTRNIGIMAHIDAGKTTTTERILYYTGRIHKIGETHEGAS 48
EFG_THETH 51 TMDFMEQERERGITITAAVTTCFWKDHRINIIDTPGHVDFTIEVERSMRV 100
.||:||||::||||||:|.||..|:.||:||||||||||||:|||||:||
EFG_STAAM 49 QMDWMEQEQDRGITITSAATTAAWEGHRVNIIDTPGHVDFTVEVERSLRV 98
EFG_THETH 101 LDGAIVVFDSSQGVEPQSETVWRQAEKYKVPRIAFANKMDKTGADLWLVI 150
||||:.|.|:..|||||:|||||||..|.||||.|.|||||.||:....:
EFG_STAAM 99 LDGAVTVLDAQSGVEPQTETVWRQATTYGVPRIVFVNKMDKLGANFEYSV 148
EFG_THETH 151 RTMQERLGARPVVMQLPIGREDTFSGIIDVLRMKAYTYGNDLGTDIREIP 200
.|:.:||.|....:|||||.||.|..|||::.||.:.|.|||||:|.||.
EFG_STAAM 149 STLHDRLQANAAPIQLPIGAEDEFEAIIDLVEMKCFKYTNDLGTEIEEIE 198
EFG_THETH 201 IPEEYLDQAREYHEKLVEVAADFDENIMLKYLEGEEPTEEELVAAIRKGT 250
|||::||:|.|....|:|..|:..:.:|.|||..||.:..||..|||:.|
EFG_STAAM 199 IPEDHLDRAEEARASLIEAVAETSDELMEKYLGDEEISVSELKEAIRQAT 248
EFG_THETH 251 IDLKITPVFLGSALKNKGVQLLLDAVVDYLPSPLDIPPIKG---TTPEGE 297
.:::..||..|:|.|||||||:||||:||||||||:.||.| :.||.|
EFG_STAAM 249 TNVEFYPVLCGTAFKNKGVQLMLDAVIDYLPSPLDVKPIIGHRASNPEEE 298
EFG_THETH 298 VVEIHPDPNGPLAALAFKIMADPYVGRLTFIRVYSGTLTSGSYVYNTTKG 347
|: ...|.:...||||||:|.|||||:|||.||||||:||||||.|:|||
EFG_STAAM 299 VI-AKADDSAEFAALAFKVMTDPYVGKLTFFRVYSGTMTSGSYVKNSTKG 347
EFG_THETH 348 RKERVARLLRMHANHREEVEELKAGDLGAVVGLKETITGDTLVGEDAPRV 397
::|||.|||:||||.|:|::.:.:||:.|.||||:|.|||||.||... :
EFG_STAAM 348 KRERVGRLLQMHANSRQEIDTVYSGDIAAAVGLKDTGTGDTLCGEKND-I 396
EFG_THETH 398 ILESIEVPEPVIDVAIEPKTKADQEKLSQALARLAEEDPTFRVSTHPETG 447
||||:|.|||||.:::|||:||||:|::|||.:|.||||||...|..|||
EFG_STAAM 397 ILESMEFPEPVIHLSVEPKSKADQDKMTQALVKLQEEDPTFHAHTDEETG 446
EFG_THETH 448 QTIISGMGELHLEIIVDRLKREFKVDANVGKPQVAYRETITKPVDVEGKF 497
|.||.|||||||:|:|||:|:||.|:.|||.|.|:||||......|:|||
EFG_STAAM 447 QVIIGGMGELHLDILVDRMKKEFNVECNVGAPMVSYRETFKSSAQVQGKF 496
EFG_THETH 498 IRQTGGRGQYGHVKIKVEPLPRGSGFEFVNAIVGGVIPKEYIPAVQKGIE 547
.||:|||||||.|.|:..|...|:||||.|||||||:|:||||:|:.|::
EFG_STAAM 497 SRQSGGRGQYGDVHIEFTPNETGAGFEFENAIVGGVVPREYIPSVEAGLK 546
EFG_THETH 548 EAMQSGPLIGFPVVDIKVTLYDGSYHEVDSSEMAFKIAGSMAIKEAVQKG 597
:||::|.|.|:|::|:|..|||||||:|||||||||||.|:|:|||.:|.
EFG_STAAM 547 DAMENGVLAGYPLIDVKAKLYDGSYHDVDSSEMAFKIAASLALKEAAKKC 596
EFG_THETH 598 DPVILEPIMRVEVTTPEEYMGDVIGDLNARRGQILGMEPRGNAQVIRAFV 647
|||||||:|:|.:..|||||||::||:.:|||::.||||||||||:.|:|
EFG_STAAM 597 DPVILEPMMKVTIEMPEEYMGDIMGDVTSRRGRVDGMEPRGNAQVVNAYV 646
EFG_THETH 648 PLAEMFGYATDLRSKTQGRGSFVMFFDHYQEVPKQVQEKLIKGQ 691
||:|||||||.|||.|||||::.|:||||.||||.:.|.:||..
EFG_STAAM 647 PLSEMFGYATSLRSNTQGRGTYTMYFDHYAEVPKSIAEDIIKKNKGE 693