Plasmids
autonomously replicating circular minichromosomes
most derived from drug-resistant bacteria
have origin of replication for amplifying DNA and marker gene to select for
cells carrying it
Modern plasmids have variety of features to make life easier.
Usually Ampicillin resistance gene, beta lactamase, as selectable marker.
Polylinker sequence - synthetic sequence with many restriction endonuclease
cut sites, so you can always find one that will work for you
Beta galactosidase gene fragment- polylinker is attached in frame to a portion
of the lacZ gene, rest is supplied by host cells.
Plasmids without inserts produce beta-gal and appear blue on X-gal plates. Insert DNA disrupts open reading frame -> no beta gal -> white colonies
Necessary because plasmid vector will recircularize efficiently if cut with
a single restriction endo.
Alternatively, one can use alkaline phosphatase to remove 5' PO4 from vector
(but not the insert), or vector can be cut with two different rest. endos. Both
of these will inhibit recircularization. Excess of insert is desirable also.
Many plasmids have promoter sequences for bacteriophage RNA polymerases flanking
the polylinker. Can be used to make strand-specific probes of insert from either
direction
Many include M13 bacteriophage origin of replication. Allows production of single strand DNA using helper phage. Useful for DNA sequencing or making site-directed mutants.
Plasmids introduced into cell by transformation:
Cells permeablized by osmotic shock or electrochemical pulse.
Host cell usually defective for restriction modification system, so exogenous
DNA not digested.
Often is mutant for several genes required for genetic recombination; this helps
in maintaining insert DNAs which might contain repeated sequences.
Other vectors:
Bacteriophage:
Lambda phage has 48.5 kb genome, packages one headful per virus particle. Can
remove middle third of genome to allow insertion of up to 16 kb of insert
Convenient for construction of Genomic and cDNA libraries

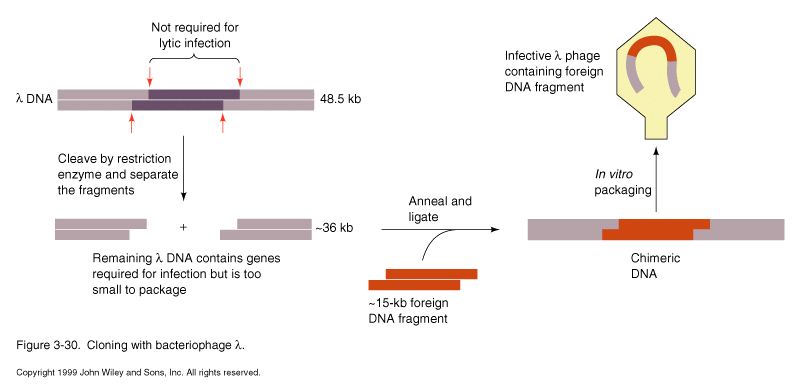
Cosmids- hybrid of lambda and plasmid, contains only packaging signal from lambda plus usual plasmid replication and marker genes. Can insert up to 45 kb insert using lambda packaging system to efficiently introduce large plasmid into cell. Replicates as plasmid thereafter.
M13 phage; filamentous bacteriophage with ssDNA genome. Has dsDNA intermediate form of DNA in cell. Can isolate dsDNA from infected cells for manipulation with rest. endos. Isolate ssDNA from particles released into culture media.
P1 phage: allows replication of up to 150 kb of DNA. Great for genomic libraries.
Shuttle vector
includes plasmid features necessary for replication in E. coli, for ease of manipulation, but also origin of replication and selectable markers for replication in other hosts, such as yeast, plants or mammalian cells
Expression vectors
Include promoter sequences to allow expression of mRNA and protein from insert
DNA. Often are expressed as fusion proteins, your gene is fused in frame with
coding sequence for another protein. Can make more stable protein than foreign
gene by itself. Also provides a handle for purification
ex.
| Fusion product | affinity resin |
| Maltose binding protein | starch |
| glutathione-S-transferase | glutathione |
| polyHis | Ni++ |
| protein A | IgG |
| beta-galactosidase | APTG |
For bacterial expression, common promoter systems are lac and T7 phage
But many eukaryotic proteins not properly expressed and modified (e. g. glycosylation) in bacteria, so eukaryotic expression systems are used. Common promoters are SV40 and CMV in mammalian cells. The baculovirus expression system in insect cells has been used to overproduce a variety of eukaryotic proteins in high yield. The promoter in this system is for the viral coat protein.
Reporter plasmids
Clone control region of your gene (promoter) upstream of gene encoding enzyme that is easy to monitor. Can look at effects of changes in cell environment on expression of reporter gene to infer effects on the intact gene of interest
Common reporters.
Chloramphenicol acetyltransferase (CAT) modifies chloramphenicol
Luciferase Produces light (ATP-dependent)
Beta-galactosidase hydrolyzes lactose analogs
The second two can be monitored in situ as well as in vitro
Cloning gene of interest:
Usually have to isolate gene of interest from a library of clones.
Library - representative collection of all (hopefully) DNA encoded in genome
or cDNA of all mRNAs expressed in cells.
Genomic DNA - usually partially digested with restriction endo or mechanically sheared and ligated into vector. --> overlapping clone sequences which span the genome, only a few of which contain your gene.
cDNA - copy of mRNA sequence made by reverse transcription of mRNA using retroviral reverse transcriptase. --> only exon sequences, much more convenient than genomic sequence for most genes (but may lose important regulatory sequences). Can make library of expressed genes for specific tissue.
For either type of library, you need a probe to detect the gene you want:
homologous DNA from related organism
cDNA for obtaining genomic clone (and vice versa)
antibody (need to use expression library)
oligonucleotide probe based on protein sequence
PCR product obtained from oligos based on shorter regions of homology
RFLP marker that maps near the gene you are interested in.
Need to know how many colonies (or phage plaques, which are usually easier to screen) are necessary to screen and be reasonably sure to get your clone.
to screen genomic DNA
P= 1-(1-f)N or N=ln(1-P)/ln(1-f)
where f is size of average insert/size of genome, P is probability,
and N= number of clones to screen
for 10kb insert you would need 2200 clones to screen the E. coli genome of 4720kb.
This could be done on a single small petri plate
You would need 1.4 million of these inserts to screen the human genome
this would require nearly 30 large petri plates.
(expression libraries require even more: gene has to be in proper orientation
and reading frame for detection with antibody)
The library is usually plated out and the phage are grown and harvested to amplify the library when it is first made. That way you or your colleagues can probe the same library many times. Or you can buy it from Stratagene or Clontech. But the number of plaques that were initially plated is critical, since each of the unamplified phage represent a unique clone.
The library is plated out and after plaques are seen, a nitrocellulose or nylon filter is overlaid onto the plate and marked to allow realignment of filter and plate later on. Filter is removed and treated with NaOH to lyse phage and denature DNA. The filter is washed with buffer and then pretreated with a hybridization buffer that contains carrier DNA and protein which bind nonspecifically to the filter (pre-hybridization). Radiolabeled DNA or RNA probe is then added and incubated at specific temperature and salt concentration to allow the probe to anneal specifically to homologous phage DNA in the library.
Typically one uses a radiolabeled DNA or RNA probe (more on that below). The temperature and buffer conditions used for hybridizing and washing the filters has dramatic effects on results.
Factors favoring hybridization:
Low temperature
High [salt]
Low [denaturant]
probe length
time
%GC content of probe
Tm=81+16.6log[Na+] — 0.4[%(G+C)] — 0.6 (% formamide) — 600/n —1.5(% mismatch)
where n is length of probe in bases
It is possible to favor hybridization too much and cause high back ground from
hybridization to plaques with little homology to the probe. These hybe conditions
are called "low stringency"
If stringency is too high, real signal could be washed off. This is particularly
likely if the probe is not an exact match (e. g. probing a human library with
a mouse DNA). In such cases the hybe conditions may have to be determined empirically,
usually using a Southern Blot (see below).
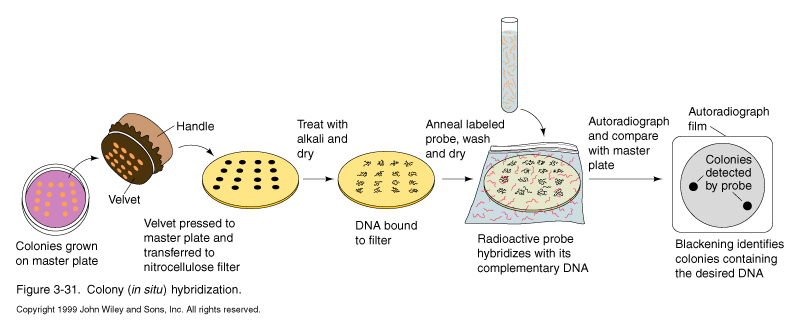
Once the filters are probed, washed and dried, they are exposed to film. Black spots indicated putative clones of interest. The film is aligned with the plate and the area around the spot it cut out phage are harvested and rescreened. Since the phage diffuse out on the plate during the hybe procedure. A couple of rounds of purification are needed to isolate pure phage cultures.
Once isolated, clones must be tested to determine whether they represent the gene of interest. One common test is the Northern blot to determine whether the DNA is homologous to a mRNA in the cell type of interest.
If you know you need to isolate more flanking genomic sequence, must do "Chromosome walking".
Make probe from the far end of your genomic clone, rescreen library.
Clones that hybridized to this probe but not the original one represent DNA further away from the original probe. Can go through multiple "steps". The larger the size of inserts, the fewer rounds you need to cover a given distance, hence the demand for cosmids, P1 vectors, and YACs
Turning expression of genes on or off in response to environmental or developmental cues. Allows adaptation to environment without producing proteins that are not needed.
E. coli lac operon
controls expression of genes required for utilization of lactose as an energy source. Genes are turned off when other sources of energy such as glucose are readily available or when lactose is absent.
b -galactosidase reaction:
b -galactosidase 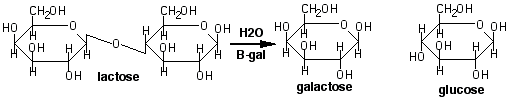
is encoded by the lacZ gene along with a cluster of related genes (an operon). lacY codes for a permease which transports lactose into the cell. lacA codes for a transacetylase of unknown function. All three genes are transcribed from a single promoter whose activity is regulated by the lac repressor (lacI).
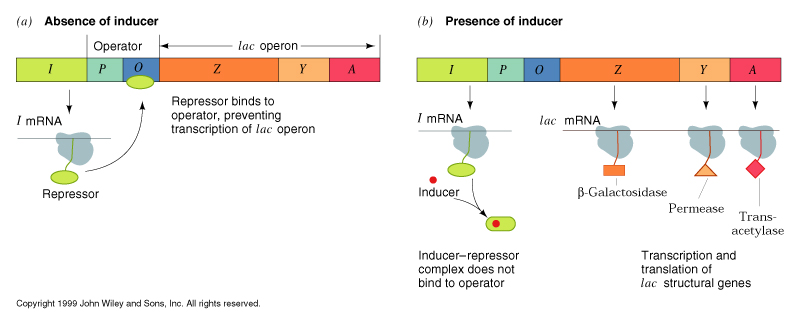
In the absence of lactose, the lac repressor binds to the operator sequence adjacent to the promoter. This inhibits transcription of the lac operon and only minute amounts of b -galactosidase are present in the cell (negative regulation). When lactose is present, the b -galactosidase, in addition to hydrolyzing lactose, will sometimes transacetylate it instead, forming 1,6-allolactose. The allolactose binds to the lac repressor, causing it to release from the operator sequence. This allows transcription of the operon to begin. Allolactose is called an inducer of the operon.
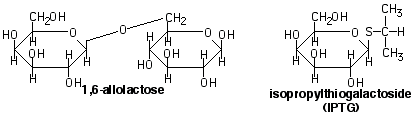
When the available lactose and allolactose are hydrolyzed, inducer concentrations fall, leaving the lac repressor free to bind the operator again and shut off the operon. In the laboratory, an analog of allolactose, IPTG, is used to induce the lac operon. IPTG binds to the repressor, but is non hydrolyzable, so operon is always on in the presence of IPTG.
The relative affinity of the lac repressor for operator DNA vs. other DNA in the presence and absence of the inducer controls the specific binding of the repressor to the operator:
|
DNA |
Repressor KD |
(Repressor + Inducer) KD |
|
lac operator |
5X10-14 M |
5X10-11 M |
|
other DNA |
5X10-7 M |
5X10-7 M |
|
Specificity |
107 |
104 |
The repressor has higher affinity for the operator than other DNA sequences even in the presence of the inducer, but binding inducer reduces affinity for the operator 1000-fold.
The lower affinity for non operator DNA is still sufficient to bind >99.99% of the free repressor, so virtually all repressor is bound to DNA. The repressor appears to bind nonspecific DNA and then track along it in a 1-dimensional search for the operator sequence. This allows repressor to bind the operator more rapidly than the diffusion controlled rate for binding directly from solution.
Lac Repressor Structure
The crystal structure of the repressor was solved in 1996. It consists of 5 domains:
Each repressor dimer can bind an operator sequence, which is a 35 bp imperfect palindrome. When the repressor core binds inducer, the conformational change is conducted through hinge to the DNA binding domains, causing them to separate, so they can no longer bind DNA simultaneously.
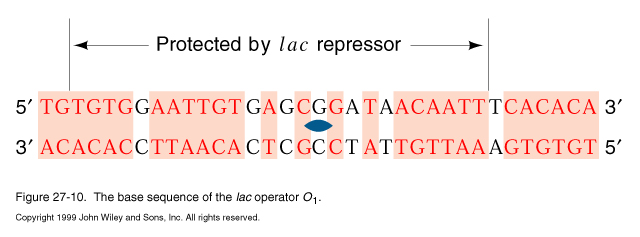
The operator region O, shown in the genetic diagram above, was defined by genetic analysis of lac mutants. It is located near the transcription start site. When the lac DNA sequence was determined, two additional copies of the operator were identified, O2 is located 401 bp into the lacZ coding sequence, O3 is located 93 bp upstream. The original operator is called O1.
Thus the repressor tetramer can bind to 2 operator sequences simultaneously, forming a DNA loop.
CAP protein
The lac operon is also positively regulated by the Catabolite Activator Protein (CAP) also called the cAMP receptor protein (CRP). Glucose is the preferred carbon source for E. coli, so the CAP regulation system turns on genes for utilization of lactose and arabinose (the ara operon), only when the amounts of glucose available are low. cAMP levels rise when glycolysis slows down in response to low levels of glucose. Thus:
¯ glucose ® cAMP ® cAMP· CAP complex ® transcription of lac, ara, etc.
The cAMP· CAP complex may act by direct contact with RNA polymerase or by altering the DNA conformation at the promoter site.
trp operon
the trp operon encodes a series of enzymes required for tryptophan biosynthesis. These genes are turned off when tryptophan is available from the environment, but are induced when trytophan is scarce and must be synthesized de novo.
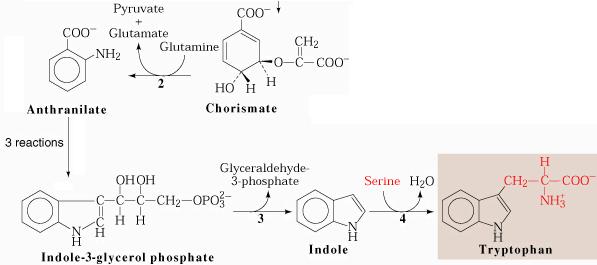
the trp synthetic pathway
Like the lac operon, the trp operon is regulated by a repressor. When the trp repressor binds tryptophan it increases its affinity for the trp operator, and transcription of the trp operon is decreased 70-fold. Tryptophan is termed a co-repressor of the trp operon.
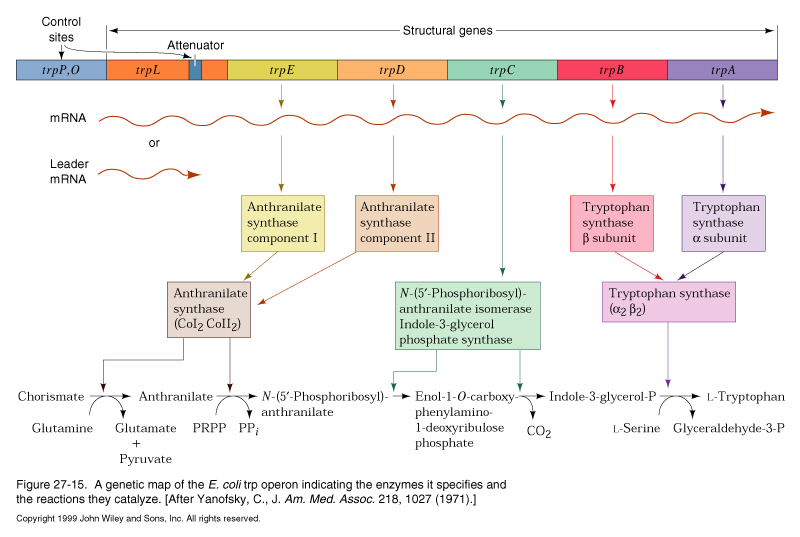
Attenuation
The trp operon has an additional level of transcriptional control. Upstream of the genes encoding the trp enzymes is a 162 nt long mRNA leader sequence which encodes a 14 amino acid peptide called the leader peptide. Immediately following the leader peptide sequence is a region of RNA which can form two alternative secondary structures:
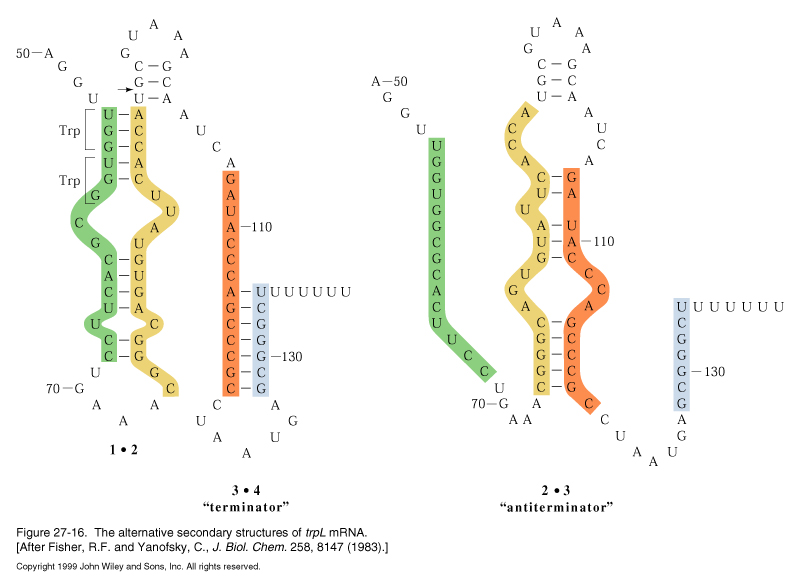
transcription into this region normally progresses up to nt 92 (arrow) and regions 1 and 2 base pair. This secondary structure causes the RNA polymerase to pause and translation begins on the nascent mRNA. The mRNA contains tandem trp codons at the beginning of region 1. If trp is abundant, the ribosome continues translating through these codons behind RNA polymerase, disrupting the 1-2 hairpin and masking it while the 3-4 region is transcribed and base pairs. The 3-4 stem followed by a string of U resides is a rho-independent termination signal and transcription by RNA polymerase terminates.
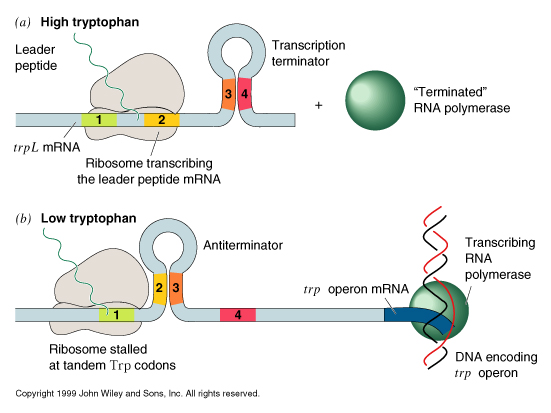
When trp levels are low, the ribosome will stall at the trp codons, due to lack of charged tRNAtrp. This masks region 1, allowing region 2 to pair with region 3 when it is transcribed. Formation of the 2-3 stem precludes formation of the 3-4 terminator, so transcription continues into the coding region for the trp biosynthetic enzymes.
Attenuation is used in other amino acid operons:
Histidine biosynthesis: his operon - 7 tandem his codons
Isoleucine, leucine and valine biosynthesis: ilv operon — 5 ile, 3 leu, 6 val codons
Bacteriophage lambda
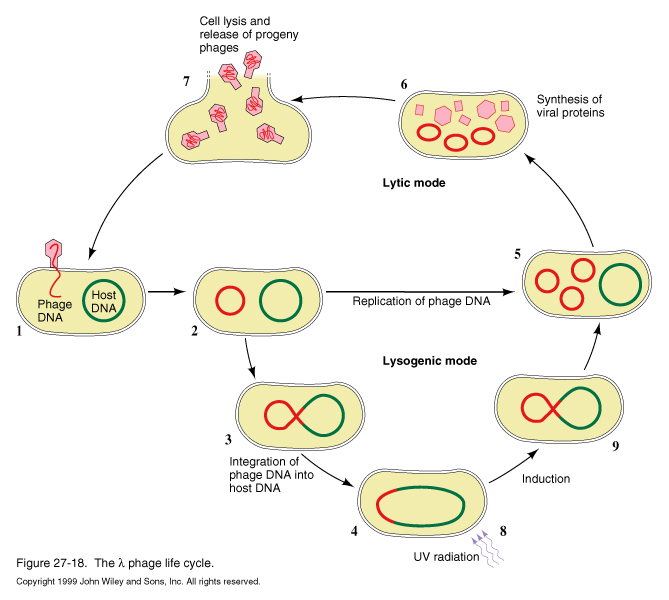
Phage can undergo lytic mode, producing burst of progeny and lysing host cell, or it can switch to lysogenic mode, integrating its DNA into the host genome, where it can remain for generations. Based upon environmental cues, the lysogenic phage can be induced to switch to lytic replication.
For either mode of replication, after infection of the host cell with phage DNA, the linear genome spontaneously cyclizes, due to its cohesive ends, which are repaired by the host ligase.
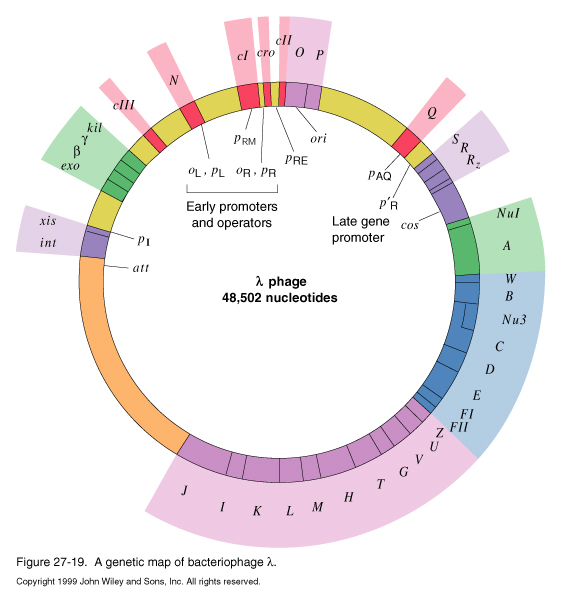
After the genome circularizes, transcription begins. The transcription pathway utilizes a variety of regulation strategies in a cascade of transcription of different operons.

The first transcripts produced are L1, R1, R2 and R4.
R1 encodes the Cro protein, which turns off synthesis of early genes.
R2 is caused by partial readthrough of the terminator for R1. Like R1, it encodes cro, but in addition, it encodes cII, O, and P genes.
The cII protein is the key factor in the decision to go through lysis or lysogeny.
O and P are necessary for DNA replication.R4 produces no protein product at early times or infection.
L1 encodes the N protein antitermination factor. N protein binds to RNA polymerase when it transcribes the nutL and nutR sequences. Binding of the N protein to RNA polymerase allows the polymerase to read through the tL1, tR1 and tR2 termination signals, allowing genes downstream of the terminators to be expressed in the next level of the gene expression cascade.
The transcripts produced by N-activated RNA polymerase are L2 R3, and R4
L2 encodes cIII protein, involved in the lysis/lysogeny decision, and the int and xis proteins required for integration and excision of the phage DNA into the host genome in lysogeny.
R3 encodes the cro, cII, O, P, and Q genes.
Q is an antiterminator, analogous to the N protein.
Late in the lytic transcription pathway, Cro protein levels are high enough to repress L1, L2, R1, R2, and R3. Q protein binds to RNA pol at the qut site, allowing synthesis of the R5 transcript, encoding genes required for viral coat proteins.
Lysogeny
The cII protein is used as an environmental sensor for the infecting bacteriophage to decide whether to proceed with the lytic pathway, or to integrate its DNA into the host genome at a specific site and follow the lysogenic pathway, replicating with the host genome.
High levels of CII protein favor lysogeny by stimulating transcription of the lambda repressor (cI gene) and int gene required for integration. Factors affecting cII levels are the multiplicity of infection (if multiple copies of phage infect a single cell, cII transcription is elevated) and the metabolic state of the host cell. In rich media, cellular proteases are active and tend to degrade the cII protein (even in the presence of cIII, which protects cII). Starved cells have low levels of proteases, resulting in high levels of cII and lysogeny.
Once lysogeny is established, it is maintained very stably until environmental cues, such as the SOS response, signal the beginning of lytic growth. The frequency of switching to lysis under inappropriate conditions is only 1 in 105, so the "genetic switch" which determines lysis vs lysogeny is a highly accurate one.
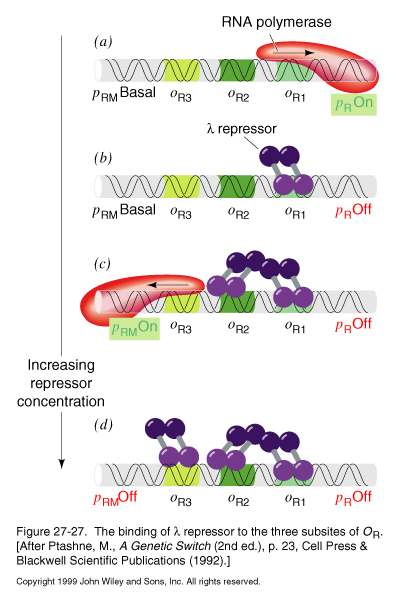
The core of the genetic switch is the interplay between two proteins, the lambda repressor and Cro, and the operator region to which they bind, oR :

oR. lies between pR (rightward promoter), which is the promoter for transcription of cro and other early lysis genes, and pRM (promoter for repressor maintainance), which is the promoter for the lambda repressor.
oR. consists of three subsites: oR1, oR2, and oR3
Cro and the repressor differ in their affinity for the different subsites.
The relative preference of the lambda repressor for the subsites is:
oR1> oR2> oR3
In addition, the repressor is a dimeric protein, with N-terminal DNA binding domains, and C-terminal domains that interact to for dimers. The C-terminal domains of different dimers can also interact with each other to allow for cooperative binding to the operator region.
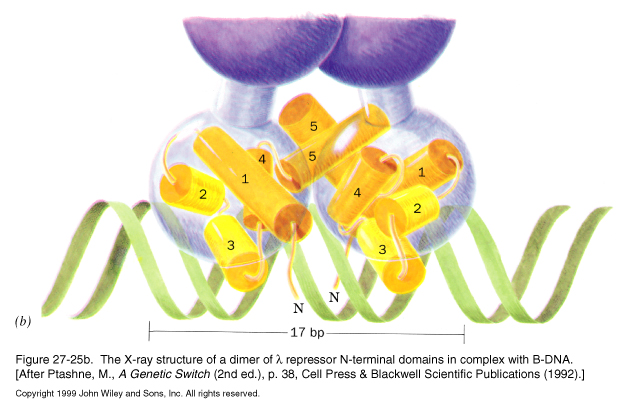
The relative affinities of the Cro protein for the operator sites are opposite those of the repressor:
oR3> oR2Å oR1
The Cro protein is also dimeric, but it lacks a domain for higher order binding, so each dimer binds each operator site independently of the other.
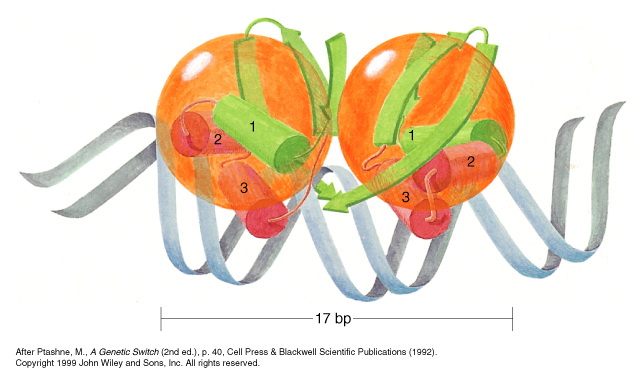
Both the repressor and Cro have helix-turn-helix DNA binding motifs, but the secondary structure of the rest of the domain differs between the two proteins.
Repressor Binding
|
|
In the absence of repressor, pR is on, leading to synthesis of cro and lysis genes |
|
At low levels of repressor concentration, the repressor dimer tends to bind oR1 first, this block transcription of pR and shuts off cro and the lysis genes. |
|
|
Binding to oR1allows for cooperative binding to oR2 , which stimulates transcription of pRM, which leads to further repressor synthesis. |
|
|
At high levels of repressor, oR3 is bound, shutting off its own synthesis by blocking transcription from pRM. |
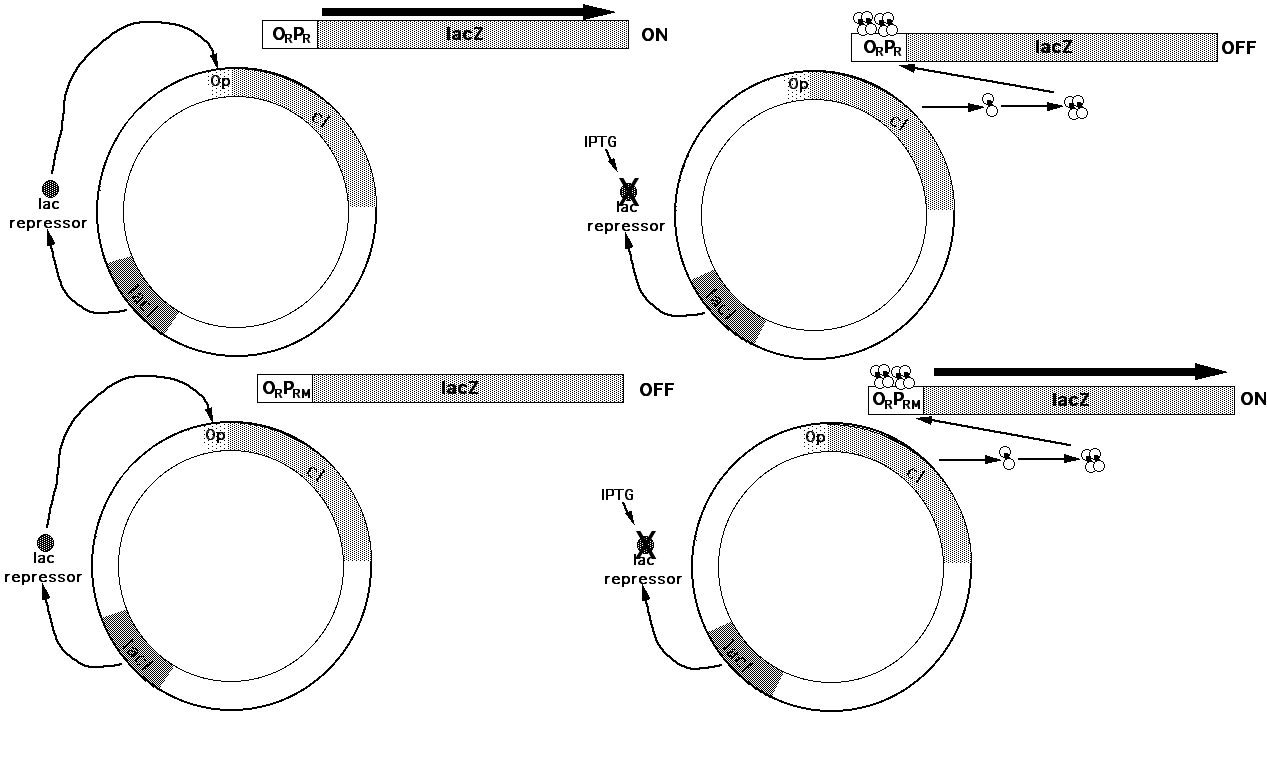
Reporter Plasmids
The lambda repressor is involved in regulating its own synthesis as well as repressing synthesis of other genes. The steps involved in this regulation were dissected by the use of b -galactosidase reporter plasmids to monitor the activity of the ORPR and ORPRM operators. The level of repressor was controlled by expressing the cI gene using the lac operator. Thus the amount of repressor protein in the cell could be adjusted by adjusting the amount of IPTG given to the cells. The effect of various levels of lac repressor on the ORPR and ORPRM operators could be easily monitored by measuring the amount of b -galactosidase activity expressed from the promoters in an in vitro b -galactosidase assay of cell extracts:
Cro protein binding
Cro binds to oR3 first, shutting of pRM, while pR remains on
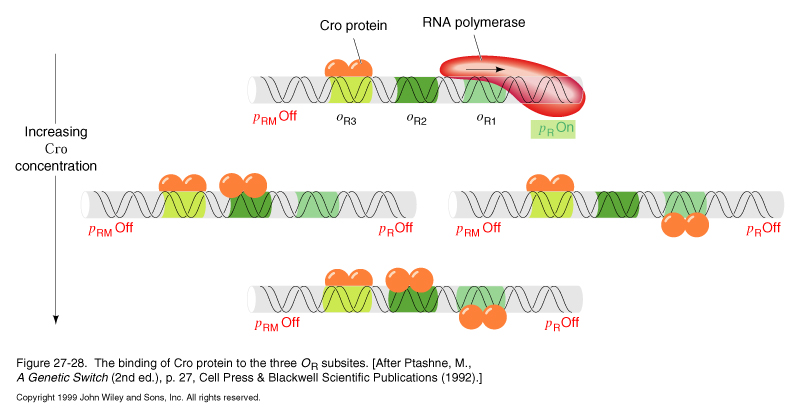
At higher concentrations of Cro, oR1 and oR2 are bound in random order without cooperativity. Binding to oR1 shuts off pR and further Cro synthesis.
Induction of lysis
The SOS response triggers protease activity of the recA protein, which attacks a number of specific targets including the lambda repressor. RecA cleaves the linker region between the N-terminal DNA binding domain of the repressor and the C-terminal domain necessary for dimerization and cooperativity. This lowers the net affinity of the repressor for the operator, causing its release and induction of the lytic pathway.