Biochemistry 601
September 8, 1999 The Process of Protein Folding
Last modified 8/23/99
Dr. Landry
Rm. 6055
landry@mailhost.tcs.tulane.edu
Bibliography:
-
Dill, K. A. (1990). Dominant Forces in Protein Folding. Biochem.
29,
7133-7155.
-
Bowie, J. U., Reidhaar-Olson, J. F., Lim, W. A., and Sauer, R. T. (1990).
Deciphering the Message in Protein Sequences: Tolerance to Amino Acid Substitutions.
Science
247, 1306-1310.
-
Noiva, R., and Lennarz, W. J. (1992). Protein Disulfide Isomerase - A Multifunctional
Protein Resident in the Lumen of the Endoplasmic Reticulum. J. Biol.
Chem. 267, 3553-3556.
-
Schmid, F. X. (1995). Protein folding: Prolyl isomerases join the fold.
Curr.
Biol. 5, 993-994.
-
Miranker, A., Radford, S. E., Karplus, M., and Dobson, C. M. (1991). Demonstration
by NMR of Folding Domains in Lysozyme.
Nature 349, 633-636.
-
Miranker, A., Robinson, C. V., Radford, S. E., Aplin, R. T., and Dobson,
C. M. (1993). Detection of Transient Protein Folding Populations by Mass
Spectrometry. Science 262, 896-900.
-
Bukau, B., and Horwich, A. L. (1998). The Hsp60 and Hsp70 Chaperone Machines.
Cell
92, 351-356.
Objectives
-
See that the stability of the native
protein fold results from the balance of large opposing forces. Net stabilization
is small.
-
Note that mutant proteins typically have
native-like structure but are destabilized relative to the "wild type"
protein.
-
Realize that protein folding must follow pathways.
-
Recognize that the efficiency of protein folding can be increased
by blocking the aggregation of folding intermediates. This is the role
of molecular chaperones (a.k.a.
heat
shock proteins).
-
Learn that the rate of folding can be accelerated by two types of
enzyme: peptidyl-prolyl cis/trans isomerase
and protein disulfide isomerase.
Native State Stabilization
The net stabilization of the native state conformation of a protein results
from the balance of large forces that favor both folding and unfolding.
For a theoretical protein, the energetic contributions to native state
stabilization may be distributed as follows:
Folding Unfolding
hydrophobic collapse conformational entropy
intramolecular H-bonding H-bonding to solvent (water)
van der Waals interactions
Contributions to the Free Energy of the reaction, U-->N:
-200 kCal/mole +190 kCal/mole
Thus the net free energy of folding is 10 kCal/mole.
-
hydrophobic collapse
-
hydrophobic sidechains coalesce in the interior of the protein structure
[Much of the free energy in this term is entropic in nature. The molecular
explanation goes as follows: solvent-exposed surface area is reduced and
thus fewer water molecules must be ordered]
The folding behavior of proteins is well approximated by a heteropolymer
model composed of two residue types, hydrophobic and hydrophilic, in a
"poor" solvent.

-
van der Waals interactions
-
weak dipole-dipole interactions between closely packed molecules
-
conformational entropy
-
entropy associated with the multiplicity of conformational states of the
disordered polypeptide chain
Interactions stabilizing the native state are cooperative
and redundant
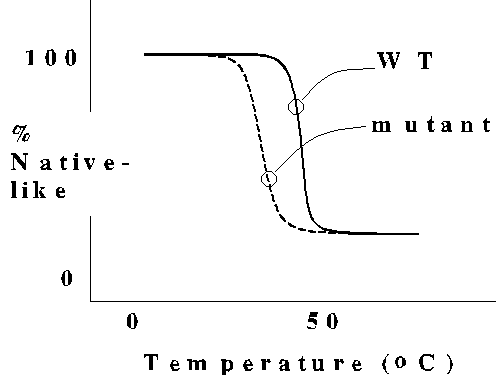
The thermal melting behavior of protein structure indicates a "catastrophic"
transition to the unfolded state, a characteristic of highly cooperative
systems. A typical point mutant causes little or no change in the structure
of the protein, but reduces protein stability as indicated by the lower
melting temperature. Note that the mutant still undergoes cooperative unfolding.
[Many amino acid substitutions in protein sequences have no detectable
effect on protein function. Mutations causing functional defects usually
occur in an enzyme active site or interfere with intermolecular assembly
(e.g., collagenopathies).]
Hemoglobin S
Note in the kinemage
that the structure of hemoglobin S is the same as normal hemoglobin, except
for the substitution of Val for Glu at position 6. Aggregation of hemoglobin
S into rod-like structures distorts the red blood cells into their characteristic
"sickle" shape.
Sequence Specifies Structure
Protein folding can be studied in vitro by unfolding the protein
with high concentrations (8 M) of a chaotropic agent such as guanidine-HCl
or urea, and then permitting the protein to refold by removal of the chaotrope
by dialysis or dilution. [Note the structural similarity of chaotropic
agents and the peptide unit. Remember the old saying, "Like dissolves like"?
The polypeptide tends to maximize its interaction with a "good" solvent
by unfolding.]

In the classic refolding experiments by Anfinsen, it was shown that
the information specifying the active conformation of ribonuclease A is
contained in the amino acid sequence. Since the native state was achieved
by either of two refolding paths, the native state lies at a global
free energy minimum.
Folding Follows Pathways
The Levinthal Paradox
Consider how long it would take a 100-residue polypeptide to complete a
random search for the native state.
Assume there are three (3) possible conformational states for each residue
and that it takes 10**(-13) sec to interconvert between each state. For
the 100-residue polypeptide, there are 3**100 [or 5x10**47] possible conformational
states. Assuming a single unique native state conformation, it would take
(5x10**47)(10**(-13)) sec = 5x10**34 sec or 1.6x10**27 yr. This absurd
result clearly shows that protein folding does not occur by random search.
Recent in vitro studies demonstrate that protein folding follows
a path(s) characterized by retention of partially correct intermediates.
Molecular Chaperones Block Aggregation of Folding
Intermediates
The efficiency of protein folding can be compromised by aggregation of
folding intermediates that have exposed hydrophobic surfaces. Such aggregates
are essentially irreversible. Molecular chaperone proteins bind
reversibly to folding intermediates, prevent aggregation, and promote their
passage down the productive folding path. Molecular chaperones also are
known as heat shock proteins because they are synthesized in much
greater amounts by cells subjected to a wide variety of stresses, including
elevated temperature and oxidative stress.
Specialized Enzymes Catalyze Key Steps in Protein
Folding
Peptidylprolyl cis/trans isomerase (PPIase)
PPIase catalyzes the cis/trans isomerization of X-pro peptide bonds,
where X is any amino acid. As previously discussed, the
partial
double bond character of the peptide bond restricts its rotation to
values that keep the atoms bonded to the CO and N in a plane. The bond
may adopt either the cis or trans conformation while satisfying
this requirement. For all but X-pro peptide bonds, the trans conformation
is strongly favored by steric
interference in the cis configuration. For X-pro peptide bonds,
a similar level of steric interference occurs in both cis and trans
conformations; thus, the cis conformer is more favored for X-pro
bonds than for bonds between other pairs of amino acids.
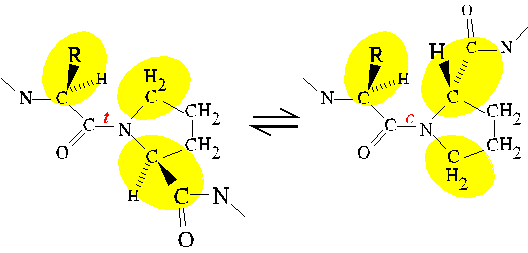
Steric interference between neighboring residues in the amino acid sequence
is an example of a local interaction. The conformational behavior
of an unfolded polypeptide is dominated by local interactions; whereas,
long-range
interactions stabilize secondary and tertiary structure in native proteins.
In an unfolded polypeptide, a given peptide bond is much more likely to
be trans (96%) than cis (4%); except in the case of an X-pro
peptide bond where the likelihood of the
cis conformer is significantly
increased (%20). This relatively high probability of cis presents
a significant barrier to the folding of a protein for which the native
secondary and tertiary structure demands the trans conformation.
Futhermore, some proteins contain a cis X-pro bond in the native
structure, so molecules with a trans X-pro bond must interconvert
to cis in order for the protein to complete folding. PPIase catalyzes
cis/trans interconversion, achieving as much as a 300-fold rate
enhancement.
Cyclophilin and FK506-binding Protein are PPIases
Immunosuppression by cyclosporin results from the formation of a ternary
complex of cyclosporin, cyclophilin, and calcineurin. Cyclophilin was named
in recognition of its affinity for cyclosporin, and it was later identified
as a PPIase. Indeed, the PPIase activity of cyclophilin is inhibited by
cyclosporin. However, immunosuppression results from inhibition of the
protein phosphatase calcineurin which is tightly bound by the cyclophilin/cyclosporin
complex. FK506 is another immunosuppressant which is chemically unrelated
to cyclosporin. FK506 is bound by FK506-binding protein (another PPIase)
resulting in formation of a calcineurin inhibitor.
Protein Disulfide Isomerase (PDI)
Disulfide bonds form and break by an exchange mechanism. Free thiol groups
of cysteine residues in proteins are oxidized to form disulfide bonds by
the reduction of preexisting disulfide bonds. Intracellularly, protein
thiols may be oxidized initially by the specialized redox molecule, glutathione
(a cysteine-containing tripeptide). The first exchange reaction between
glutathione dimer (G-S-S-G) and the protein yields a mixed disulfide. The
mixed disulfide then undergoes an intramolecular exchange reaction resulting
in the discharge of reduced glutathione and formation of the intrachain
disulfide bond. The polypeptide may then isomerize through the exchange
of disulfide bonding partners. [In Anfinsen's experiment, "scrambled" ribonuclease
isomerized to the native disulfide pattern after addition of a small amount
of reduced beta-mecaptoethanol to break one of the disulfide bonds.] In
cells, these isomerization reactions are catalyzed by PDI. The mechanism
of PDI catalysis involves the transient formation of a disulfide bond between
PDI and the polypeptide.

End of document