(African sleeping sickness)
Trypanosoma cruzi
(Chagas' disease)
Leishmania
species
(leishmaniasis)
| Kinetoplastids Causing Human Disease |
|---|
| African
trypanosomes (African sleeping sickness) Trypanosoma cruzi
Leishmania
species |
|
|
|
The kinetoplastids are a widespread group of flagellated protozoa. Members of this group parasitize virtually all animal groups as well as plants and insects. There are also free-living kinetoplastids which feed on bacteria in aquatic, marine and terrestial environments. The kinetoplastids are likely a monophyletic group which are related to the euglenids. Three distinct kinetoplastids cause human disease: African typanosomes (African sleeping sickness), Trypanosoma cruzi (Chagas' disease), and Leishmania species (leishmaniasis). All three are parasites of the blood and/or tissues of the human host and are transmitted by arthropod vectors (see page on vectors). (Link to another page on kinetoplastids focusing on African trypanosomes.)
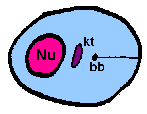
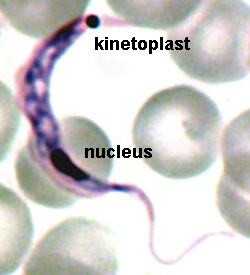
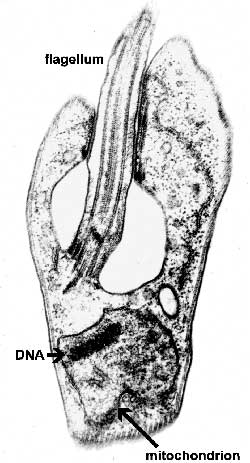
The major distinguishing feature of this group is a subcellular structure known as the kinetoplast. The kinetoplast is a dark Giemsa-staining structure which is distinct from the nucleus (Figure). The size of the kinetoplast will vary according to species. The kinetoplast is found near the basal body which is located at the base of the flagellum (Figure). Because of this location near the flagellum, it was previously believed that the kinetoplast was somehow associated with cell movement--hence the name. However, the kinetoplast is actually a distinct region of the mitochondria and is not involved in motility. The staining of the kinetoplastid is due to mitochondrial DNA (see Box). In fact, the existence of extranuclear (i.e., organellar) DNA was first demonstrated in the kinetoplastids.
|
Kinetoplastid DNA Kinetoplastid DNA is relatively abundant and consists of mini-circles and maxi-circles. The two types of ktDNA occur in a concatenated mass within the mitochondria. Maxi-circles encode several mitochondrial genes and are more-or-less equivalent to the mtDNA. Mini-circles are heterogeneous and rapidly evolving and their function is less clear. Both mini-circles and maxi-circles encode guide RNA genes. Some genes on the maxi-circles have 'errors' which need to be edited. The guide RNAs are important for this RNA editing that takes place in the mitochondria of kinetoplastids. The editing of these 'cryptogenes' is believed to occur in a hypothetical 'editosome' particle. The extent of editing seems to correlate with different parasite life cycle stages and the corresponding changes in metabolism (i.e., aerobic vs. anaerobic) that are associated with the different life cycle stages. Mini-circle DNA is also used for parasite detection and distinguishing different isolates. |
Typically, the kinetoplastids are depicted as long slender organisms. However, the kinetoplastids exhibit several morphological forms which are defined by the position of the kinetoplast in relation to the nucleus and the length of the undulating membrane (see morphological forms). Cellular features of the kinetoplastids include:
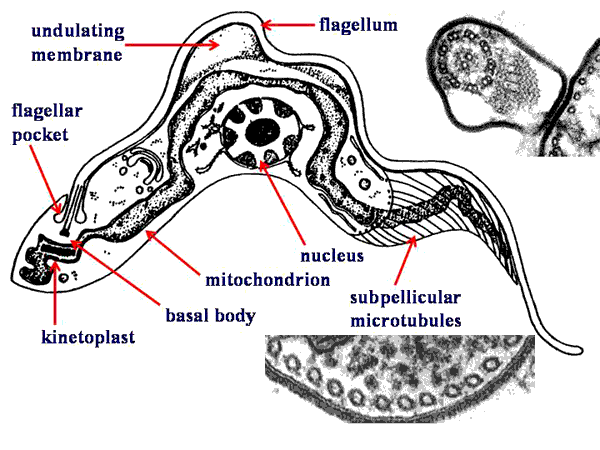
Morphological FormsSeveral different morphological forms of kinetoplastids are observed. These various morphological forms are associated with different life cycle stages in the various species. The different forms are distinguished by the position of the kinetoplastid in relation to the nucleus and the presence or absence of an undulating membrane. The four major morphological forms found in kinetoplastids which cause human disease are:
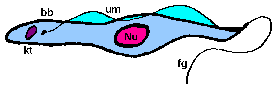
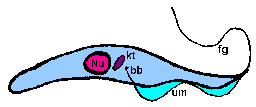
trypomastigoteThe kinetoplast (kt) is located on the posterior end of the parasite. The flagellum emerges from the posterior end and folds back along the parasite's body. This attachment of the flagellum to the body forms an undulating membrane (um) that spans the entire length of the parasite and the free flagellum emerges from the anterior end. This is considered the anterior end since the flagellum pulls the organism and the end with the free flagellum is the front in reference to the direction of movement. The undulating membrane functions like a fin and increases the motility of the organism.
epimastigoteThe kinetoplast (kt) is more centrally located, usually just anterior to nucleus (Nu). The flagellum (fg) emerges from the middle of the parasite and forms a shorter undulating membrane (um) than observed in trypomastigotes. Epimastigotes are noticeably less motile than trypomastigotes.
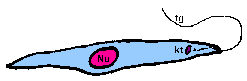
promastigoteThe kinetoplast (kt) is towards the anterior end and a free flagellum (fg) with no undulating membrane emerges. The end that the free flagellum emerges from in all three motile forms is designated as the anterior end because they swim in that direction. In other words, the flagellum pulls the organism.
amastigoteThe parasite is more spherical in shape and has no free flagellum. A basal body (bb) and the base of the flagellum is still present. The kinetoplast (kt) is usually detectable as a darkly staining body near the nucleus (Nu). This form is a non-motile intracellular stage. |
 |
 |
 |
 |
African trypanosomiasis, also known as African sleeping sickness, exhibits a patchy distribution in equatorial Africa depending upon specific topographical features and the presence of the vector. The control of African trypanosomiasis is complicated by poverty, political instability, and civil wars often found in areas endemic for the parasite and vector. An estimated 60 million people in 36 nations are at risk of infection. The incidence of the disease had been increasing from the mid-1960s to the end of the 20th century with an estimated 300,000-500,000 cases occuring annually in 1998. However, increased awareness and programs initiated by the WHO have led to a decreasing incidence and in 2009 there were less than 10,000 cases (see Simarro et al, 2011).
The parasites responsible for causing African sleeping sickness belong to a group of closely related trypanosomes in the Trypanosoma brucei species complex. Three morphologically indistinguishable species are recognized:
| T. brucei | infects game animals/livestock (causes nagana) | |
| T. rhodesiense | causes E. African trypanosomiasis | |
| T. gambiense | causes W. and Central African sleeping sickness |
(Some authors consider these as subspecies: T. brucei brucei, T. b. rhodesiense, T. b. gambiense.)
T. brucei is a natural parasite of wild game in Africa and are non-infective to humans. This inability to infect humans is due to a 'trypanosome lytic factor' found in human sera. T. brucei and two morphologically distinct trypanosmes, T. vivax and T. congolense, are major pathogens for wild and domestic animals and have far reaching effects on raising livestock. In fact, although trypanosomiasis can be a devastating human disease, the greatest impact of trypanosomiasis on human health is at the agricultural level. Large areas of Africa are unsuitable for raising cattle and other livestock due to the presence of the tsetse vector and the transmission of trypanosomes. This contributes to protein deficient diets among the indigenous population.
As the names imply, T. gambiense and T. rhodesiense are distinguished by their geographical distributions. T. rhodesiense is found in East Africa and T. gambiense is found in West and Central Africa. (Rhodesia is the former name for Zimbabwe.) The restricted distributions of the African trypanosomes are determined by the vectors. African trypanosomes are transmitted by several species within the genus Glossina, commonly known as the tsetse. (See more on tsetse vector and parasite-vector interactions.) In addition to being transmitted by different vectors, T. gambiense and T. rhodesiense are distinguished by animal reservoirs, epidemiology, and disease virulence (Table).
| Major Differences Between African Trypanosome Species | |||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
|
The tsetse vector of T. rhodesiense is found in woodland or dry bush environments. Its natural vertebrate host is the antelope and other wild ungulates. Disease transmission depends upon coming in contact with infected vectors and is often associated with hunters or safaris. T. rhodesiense is primarily a zoonosis and little human-fly-human transmission occurs. The disease typically progresses rapidly and is often fatal without treatment. Although variants of T. rhodesiense which cause a slowly progressing chronic disease have been identified (MacLean et al, 2004, Infect. Imm. 72,7040). Molecular data suggest that T. rhodesiense may be a host-range variant of T. brucei (see box).
T. gambiense is transmitted by vectors found along rivers and lakes which are often in close proximity to human habitation. Consequently, the disease tends to be endemic and domestic animals may serve as reservoirs. The disease is much less virulent and is characterized by a slow progression from an acute disease to a chronic disease. It is believed that T. gambiense has been associated with humans for much longer than T. rhodesisense and thus possibly accounts for the lower virulence of T. gambiense.
|
The genetic and evolutionary relationships between T. gambiense, T. rhodesiense, and T. brucei may provide some insight into the evolution of human disease. Molecular techniques show that T. gambiense is relatively homogeneous throughout its wide distribution in central and western Africa. In contrast, distinguishing T. rhodesiense and T. brucei has been much more problematic. Molecular analyses indicate that both of these species exhibit a wide range of sequence heterogeneity and in some cases more homology is observed between T. rhodesiense and T. brucei than between isolates of the same species obtained from different geographical regions. The discovery of a single gene, called SRA, that can confer resistance to trypanosome lytic factor (see more on TLF ) has provided some insight into the evolutionary relationships between T. rhodesiense and T. brucei. SRA is a truncated VSG (see antigenic variation) and is only found in T. rhodesiense. Presumably this mutation in the ancestral T. brucei conferred human infectivity and thus represented the origin of T. rhodesiense. Transfer of this gene to other T. brucei isolates with different genetic backgrounds via sexual recombination (see more on sex in trypanosomes) would account for the genetic heterogeneity of T. rhodesiense and its spread in eastern Africa. In this regard, one might also consider T. rhodesiense a host-range variant of T. brucei. W. Gibson (2002) Will the real Trypanosoma brucei rhodesiense please step forward? Tr. Parasitol. 18:486. |
|
|
|
|
|
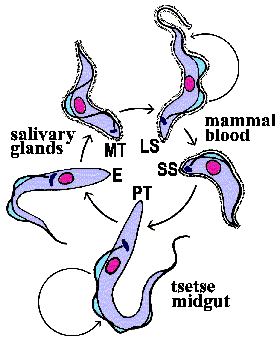 Metacyclic
trypomastigotes (MT) in the saliva of the tsetse
are transferred to the bloodstream of the mammalian host as the tsetse feeds
(Figure). The parasite exhibits a trypomastigote
morphology in the bloodstream and is extracellular. These extracellular forms
undergo an antigenic variation
to evade the host immune system. Within the bloodstream the trypanosome undergoes
asexual replication by longitudinal binary fission. These replicating forms
are generally long slender (LS) parasites. In addition to the long slender forms,
intermediate and short stumpy (SS) forms are also found within the bloodstream
of the mammalian host. The short stumpy forms are thought to be preadapted for
the tsetse. However, in vitro
experiments suggest that all bloodstream forms are infective for the tsetse.
Metacyclic
trypomastigotes (MT) in the saliva of the tsetse
are transferred to the bloodstream of the mammalian host as the tsetse feeds
(Figure). The parasite exhibits a trypomastigote
morphology in the bloodstream and is extracellular. These extracellular forms
undergo an antigenic variation
to evade the host immune system. Within the bloodstream the trypanosome undergoes
asexual replication by longitudinal binary fission. These replicating forms
are generally long slender (LS) parasites. In addition to the long slender forms,
intermediate and short stumpy (SS) forms are also found within the bloodstream
of the mammalian host. The short stumpy forms are thought to be preadapted for
the tsetse. However, in vitro
experiments suggest that all bloodstream forms are infective for the tsetse.
The tse-tse can ingest trypanosomes with its blood meal. The bloodstream trypomastigote differentiates into a procyclic trypomastigote (PT) within the gut of the tsetse. Accompanying this differentiation is a loss of the VSG surface coat and changes in the mitochondria and metabolism. The environment within the gut of fly is quite different than that of the mammalian bloodstream. The mammalian bloodstream is rich in glucose and parasite exhibits a high rate of glycolysis which is carried out in a special organelle known as the glycosome. Because of this abundance of glucose the parasite does not carry out oxidative phosphorylation within the mitochondria and consequently the mitochondria are acristate and have minimal electron transport activity. Within the vector, though, mitochondrial functions associated with aerobic metabolism return and cristae develop within the mitochondria. The procyclic trypomastigotes undergo multiple rounds of asexual replication within the midgut of the tsetse. The procyclic stage can also be cultured in vitro.
The focus in medical parasitology courses tends to be on the complex interactions between the parasite and the human host which result in pathology. However, parasites also interact with and undergo complex developmental processes in the vector. Vectors are more than 'flying syringes'. (Although T. evansi, a trypanosome infecting horses and camels, is transmitted mechanically by horseflies.) One problem for the trypanosome is that it must move from the gut to the salivary glands of the tsetse. The exact mechanism by which the parasite migrates from the tsetse gut to the saliva glands is not known. Two routes have been proposed: 1) the classical route in which the parasite 'backtracks' through the digestive system and migrates up the salivary duct, or 2) the direct route in which the parasite penetrates the peritrophic membrane and gut epithelium to gain access to the hemolymph. (Click here for more discussion on the tsetse and vector-trypanosme interactions.)
After reaching the salivary glands the procyclic trypomastigotes transform into epimastigotes (E) and attach to epithelial cells via their flagella. The epimastigotes probably undergo further replication within the salivary gland. The epimastigotes are non-infective for the mammalian host and they must first mature into metacyclic trypomastigotes (MT). During this maturation the surface coat is reformed, the mitochondria lose their cristae and the parasite detaches. These trypomastigotes are free within the lumen of the salivary gland waiting to be transferred to a vertebrate host when the tsetse feeds again, thus completing the life cycle.
Review of life cycle: Vickerman (1985) Developmental cycles and biology of pathogenic trypanosomes. Br. Med. Bull. 41, 105-114.
|
The long-standing dogma has been that trypanosomes exhibit no sexual stages. However, molecular studies (first reported in 1986) indicate that genetic recombination occurs in the African trypanosomes within the tsetse vector. Although genetic recombination clearly occurs, it is not obligatory and it is rare. It is not yet known exactly when this recombination occurs within the life cycle. Experimental evidence indicates that hybrids are only found in salivary glands and not in procyclic stages found in the gut. Most likely the fusion is between epimastigotes and not metacyclic trypomastigotes. No haploid gamete stages have been observed the biparental inheritance of the kDNA suggests that mitochondria exchange DNA. A genetic cross between T. brucei and T. rhodesiense has been successfully carried out in the laboratory. In regards to the human serum resistance phenotype (see more on TLF and its mode of action), both parental phenotypes (i.e., sensitive and resistant), as well as intermediate forms, were detected in the progeny. W. Gibson (2001) Sex and evolution
in trypanosomes. Int.
J. Parasitol. 31:643. |
|
|
|
|
|
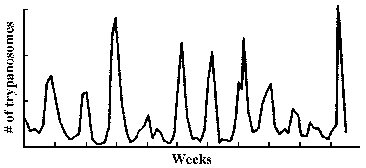
The observation that an extracellular parasite which is found in the bloodstream causes a long-lasting chronic disease poses a paradox. Normally the host immune system is quite efficient at generating antibodies against infectious organisms and eliminating them from the circulation. In this regard, a characteristic of African trypanosomiasis is a fluctuating parasitemia (Figure, upper left). In other words the number of parasites in the circulation dramatically rises and falls. Generally, fever and other clinical symptoms are associated with the peaks in parasitemia. Further examination of parasites obtained from successive peaks reveals that they are antigenically distinct, or exhibit variant antigenic types (VAT).
 |
 |
 |
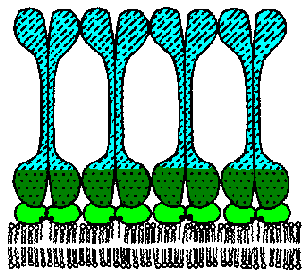
The VAT are determined by a protein known as the variant surface glycoprotein (VSG). VSG is an abundunt protein (107 copies per cell) and is a major component of the 12-15 nm thick electron dense 'surface coat' covering bloodstream trypomasigotes. The parasite is estimated to have more than 1000 distinct VSG genes (occupying ~10% of the trypanosome's genome). Periodically the parasite will express a different VSG gene which is antigenically distinct from the previously expressed VSG with a switch rate of approximately 10-2 per cell per generation. Comparing the sequences of the various VSG genes reveals a N-terminal variable domain and a C-terminal conserved domain (Figure, lower left). Also at the C-terminus is a GPI anchor which is imbedded into the lipid bilayer of the plasma membrane. The VSG molecules fold and pack together in a manner so that the conserved C-terminal region is not directly accessible and that only the varible N-terminal region is exposed to the host immune system (Figure, right).
VSG is immunogenic and antibodies against VSG do lead to parasite elimination. Switching expression to another VSG results in a new surface coat which is now not recognized by the host antibodies. These parasites will then rapidly increase in number until the host mounts an immune response against the new VSG. The large repertoire of antigenically distinct VSG proteins means that the parasite stays one step ahead of the host and avoids complete elimination by the immune system.
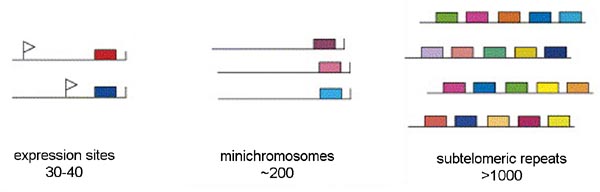
Three distinct pools of VSG genes are found in the genome of the African trypanosome (Figure). The majority of the VSG genes are found in long tandem arrays of repeated genes in subtelomeric locations on the chromosomes. Approximately 200 copies of VSG genes are found at the telomeres of the mini-chromosomes. African trypanosomes contain approximately 100 mini-chromosomes of 50-100 kilobases which only code for VSG. Another 30-40 VSG genes are found in expression sites. There are two types of expression sites corresponding to the two life cycle stages in which VSG is expressed on the parasite surface: metacyclic expression sites and blood-stream form expression sites. The blood-stream form expression sites produce a polycistronic RNA containing several genes in addition to VSG that is processed to the mature mRNA and the metacyclic expression sites only have a VSG gene.
Only a single VSG gene is expressed at a time in an individual trypanosome. The expression of a single gene appears to be regulated by an epigenetic mechanism. There is evidence suggesting that the active expression site is located in a distinct region of the nucleus and only a single expression site can occupy this location. Therefore, only a single gene can be expressed per trypanosome. There are three mechanisms for switching the VSG that is being express (Figure): in situ activation, telomere exchange, and gene conversion.
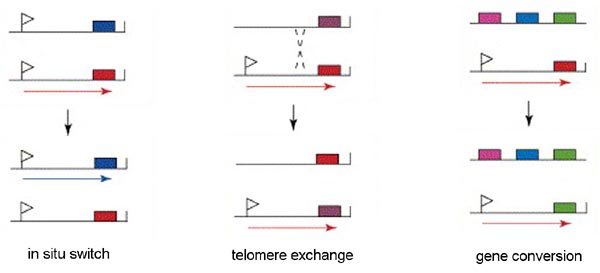
'In situ' gene activation (i.e., transcriptional control) involves turning off the active expression site and initiating expression from another expression site. This in situ switching appears to predominate early in the infection and may represent a 'preprograming' of antigenic variation. Similarly, transcriptional regulation is also associated with metacyclogenesis. A limited subset of VSG genes can be expressed by metacyclic trypomastigotes found within the salivary glands of the tsetse (see life cycle). The expression of VSG and the surface coat is a preadaptation of the parasite for the vertebrate host.
Another mechanism of VSG switching involves telomere exchange between the telomere with the active expression site and a telomere of a silent expression site or the telomere of a minichromosome. The exchange of the telomeres results in the expression of a previously silent gene and the silencing of the previously expressed gene.
The third mechanism of VSG switching involves a duplicative transposition of these VSG genes into the active expression site. The archive copy of the VSG gene is copied and the template remains intact. The copied gene then replaces the gene in the active expression site by a gene conversion process.
Sequencing of the genome of African trypanosomes revealed that relatively few (approximately 7%) of the VSG genes are intact functional genes, whereas the majority of the VSG genes have frame shift errors or in-frame stop codons. These pseudogenes undergo an intragenic recombination which can restore the coding sequence. The formation of these chimeric genes also leads to the formation of novel VSG molecules. This gives the African trypanosome an enormous potential to generate diversity. Thus the repertoire of VSG genes is continuously changing in that new variants are continuously being created whereas recently expressed variants are being lost due to duplicative transposition (i.e., gene conversion).
See also:
|
|
|
|
|
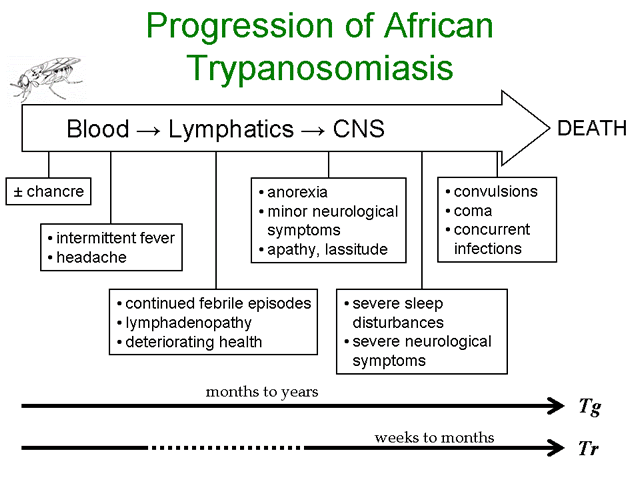
Infection with African trypanosomes can result in disease manifestations ranging from asymptomatic or mild to a severe fulminating disease. T. rhodesiense is more likely to cause a rapidly progressing and fulminating disease than T. gambiense. T. gambiense tends to cause a slow progressing disease which may either be self-limiting or develop into a chronic disease involving the lymphatics and the central nervous system (CNS). The infection is almost always fatal with few documented cases of individuals clearing the parasites and surviving.
| Disease Progression |
|---|
|
The infection is initiated when metacyclic trypomastigotes are introduced from the salivia of the tsetse into the bite wound. Generally there is an asymptomatic incubation period of 1-2 weeks in which the trypomastigotes are replicating within the tissue near the site of the bite. Occasionally, a local inflammatory nodule known as a 'trypanosomal chancre' is observed during this period. Chancres are usually tender and painful and ulceration may occur.
The trypomasigotes will invade the capillaries and enter the circulatory system during this incubation period and continue to replicate within the blood of the human host. The establishment of this acute blood stage infection is characterized by irregular episodes of fever and headache. In the case of T. gambiense the number of parasites in the blood tends to be very low and often the infected person exhibits no symptoms, whereas most persons infected with T. rhodesiense will exhibit much higher parasitemias and a more pronounced fever sometimes associated with rigor.
Disease progression is often characterized by invasion of the lymphatics in T. gambiense infections. Symptoms during the lymphatic stage include enlarged lymph nodes (particularly post-cervical group), weight loss, weakness, rash, itching, and edema as well as the continued intermittent febrile attacks. Higher parasitemias are often associated with the symptomatic periods. The infection can spontaneously resolve during either the blood stage or the lymphatic stage. There is usually little evidence of lymphatic involvement in T. rhodesiense infections. In general, the symptoms during the earlier stages of the infection tend to be non-specific (fever, malaise, headache, weakness) and may infect multiple organs.
A hallmark feature of African trypanosomiasis is the invasion of the CNS and nervous system impairment. Trypanosomes crossing the blood-brain barrier result in a generalized meningoencephalitis characterized by progressively worsening symptoms. Indications of nervous impairment include: apathy, fatigue, confusion, somnolence, and motor changes (such as tics, slurred speech, and incoordination). The changes in sleep patterns are often characterized by extreme fatigue during the day and extreme agitation at night. Generally it is 6-12 months (or even years) after the infection before the neurological symptoms start to become apparent in the case of T. gambiense. Neurological manifestations can occur within weeks after T. rhodesiense infections. If untreated, the CNS stage of the disease will almost always progress to include convulsions or coma followed by death in both T. gambiense and T. rhodesiense infections.
| Diagnosis | Treatment | Prevention |
|---|---|---|
|
|
|
Confirmed diagnosis depends upon the detection of trypanosomes in the blood, lymph node aspirations, or spinal fluid. Typically few trypanosomes are detected in the blood or other bodily fluids during the T. gambiense infections. The trypanosomes are more likely to be detected during symptomatic periods (eg., during febrile episodes). In the absence of detectable parasites, travel or residence in an endemic area combined with the symptoms discussed above can be used as a presumptive diagnosis.[Detailed descriptions of diagnostic methods are available at OIE.]
| Parasite Detection in Blood |
|---|
|
Because of the low parasitemias exhibited during T. gambiense infections it is often not possible to detect parasites by standard thin and thick blood smears. Techniques to increase the sensitivity of detection are often needed (Box). For example, looking at fresh whole blood mounts may increase the sensitivity due to the distinctive movement of the trypomastigote. Another method to increase sensitivity is to centrifuge the blood in a microhematocrit tube. The parasites are enriched in the 'buffy coat' which is the band of white cells directly about the packed erythrocytes. The tube is then broken at the buffy coat and Giemsa-stained blood smears are prepared from these cells. Inoculation of mice or rats with patient blood and looking for the development of parasitemia is also possible. This generally works much better for T. rhodesiense infections than T. gambiense infections.
The miniature anion-exchange centrifugation technique (mAECT) is another method used in the diagnosis of human sleeping sickness. Blood is passed through an anion exchange column. Blood cells are more negatively charged than the trypanosomes and they are retained on the column. The trypanosomes pass through the column and are collected at the bottom of a sealed glass tube by low-speed centrifugation. The tip of the glass tube is then examined in a special holder under the microscope for the presence of trypanosomes. The large blood volume (300 µl) enables the detection of less than 100 trypanosomes/ml. Although highly sensitive, the manipulations are somewhat tedious and time-consuming. (See http://www.finddiagnostics.org/programs/hat-ond/hat/parasite_detection/mAECT/)
One important issue in diagnosis of African trypanosomiasis is to distinguish the late encephalitic stage of the disease from the early stage. This is important since the treatment is different depending on whether there is CNS involvement (see below). Criteria for CNS involvement include detection of parasites in the cerebral spinal fluid (CSF) or elevated white blood cells (>5/microliter) in the CSF. There is some controversy in regards to the exact level of white blood cells in the CSF should constitute a classification of CNS involvement.
Suramin and pentamidine are the recommend drugs during the acute stage without CNS involvement, whereas melarsoprol or eflornithine are recommend if the CNS is involved (Table). Pentamidine is less toxic than suramin. However, it is not effective against T. rhodesiense. All four of these drugs are provided free of charge by the World Health Organization through public and private partnerships with pharmaceutical firms. The prognosis is generally excellent if treatment starts during the acute stage.
| Drug | Use | Drawbacks |
| Pentamidine | Effective against early-stage gambiense disease |
|
| Suramin | Effective against early-stage gambiense and rhodesiense disease |
|
| Melarsoprol | First line drug for late-stage gambiense and rhodesiense disease involving CNS |
|
| Eflornithine | Effective against late-stage gambiense disease involving CNS |
|
The drugs used to treat trypanosomiasis are less than ideal due to problems with toxicity and other drawbacks (Table). For example, melarsoprol, an arsenical based drug, has been used for more than 50 years despite it relatively high toxicity (up to 5% of patients die from the drug treatment). Furthermore, the treatment course is long (minimum 10 days) and requires hospitalization, thus adding significantly to the costs. There is an urgent need for the development of better anti-trypanosomal drugs. However, since the victims of African sleeping sickness are primarily the rural poor, there is little financial incentive for research and development. In fact, among the anti-trypanosomal drugs currently being used, suramin was introduced in 1920, pentamidine was introduced in 1941, melarsoprol was introduced in 1949, and eflornithine was introduced in 1990.
Eflornithine, an ornithine decarboxylase inhibitor, is a recently developed effective anti-trypanosomal drug. It has been nicknamed the ‘resurrection drug’ because of its spectacular effect on comatose patients in late stage African sleeping sickness due to T. gambiense. However, it is not effective against T. rhodesiense. Eflornithine is also expensive and the standard treatment is 14 consecutive daily injections. Oral formulations are currently being developed. For more information see TDRnews, (WHO). The most recent advance in the treatment of T. gambiense HAT has been the introduction of nifurtimox-eflornithine combination therapy (NECT).
[See a case report on two returning safari tourists with African trypanosomiasis for a description of the clinical symptoms, diagnosis and treatment of T. rhodesiense. Emerg. Inf. Dis. 8:74.]
Wearing protective clothing or using insect repellents are the recommended prophylactic measures against African trypanosomiasis. Other measures to lessen contact with the day-biting tsetse, such as avoidance of streams and water holes during the warm dry season, can also be taken. Prophylactic drugs are contraindicated since they may mask latent CNS infections and promote drug resistance. In addition, the available drugs are somewhat toxic.
Control activities are primarily focused on T. gambiense and involve reducing the number of infected humans as well as reducing the vector. Systematic surveillance and treatment of infected persons is an effective control measure since humans are the primary reservoir and the disease in the early stages is often asymptomatic or mild and slowly progressing. Control of the riverine tsetses includes destruction of their habitats and breeding places by clearing stream banks of trees and shrubs. Widespread application of insecticides can also be used to reduce the number of tsetse.
The use of traps and targets are another potential means to reduce the number of tsetse in a localized area. Traps and targets function by attracting the tsetse to a contraption that collects or kills them and thereby reduce a fraction of the tsetse population. Traps can be used for entomological surveillance, as well as for control. Targets are screens that are impregnated with biodegradable insecticides in order to kill any flies that land on them. The strategy is to place the traps or targets around a village or other areas such as along riverbanks where bathing and washing activities take place or along paths. This will lead to a reduction in the number of tsetse in that vicinity. Traps and targets are also advantageous in that there is less environmental impact than widespread insecticide spraying or clearing brush from large areas. Although this strategy is relatively low-tech, it does require community awareness and participation. The traps and targets need to be maintained and replenished with attractants and insecticides.
Reviews on African Trypanosomiasis:
|
|
|
|
|
Trypanosoma cruzi is the causative agent of Chagas' disease. Chagas' disease exhibits a patchy distribution throughout south and central America. The infective stage of the organism was discovered by chance while Carlos Chagas was studying the vectors. He named the organism after his mentor Oswaldo Cruz. Chagas determined the life cycle and found that the parasite could infect a wide range of mammals, including humans, and subsequently described the salient features of the disease. Chagas' disease is the leading cause of heart disease in south and central America.
T. cruzi is transmitted to the vertebrate host by various species of the Triatominae subfamily. The most important vectors for human transmission are Triatoma infestans and Rhodnius prolixus. Members of this subfamily are blood-feeding insects and are called many different names including: triatomine bugs, reduvid bugs, kissing bugs, cone nosed bugs and assassin bugs. While taking a blood meal the triatomines defecate and infective metacyclic trypomastigotes are released in the feces. Often the trypomastigotes will gain access to the vertebrate host by entering through the bite wound (step 1). The parasite is also capable of penetrating mucous membranes and hair follicles. The triatomines are nocturnal feeders and the bite is painless. Typically the bugs feed near the mouth or eyes of the human host. Infection is often associated with rubbing the infected fecal material into the bite wound or eyes while sleeping.
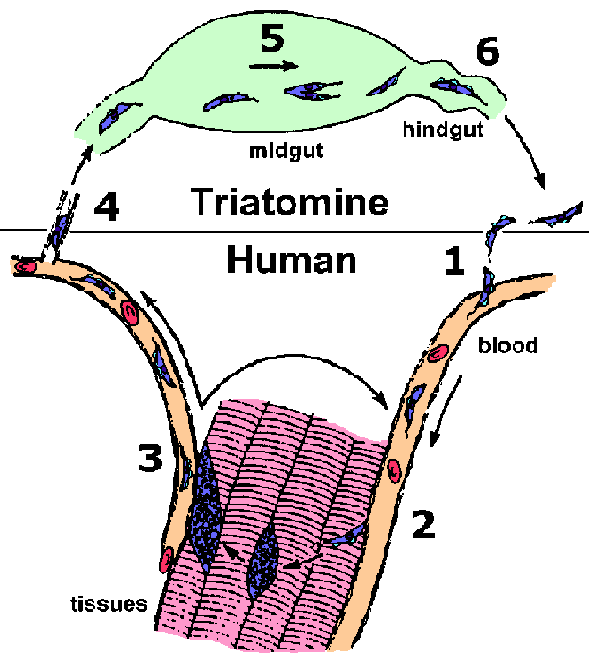
In contrast to the African trypanosomes, the trypomastigotes of T. cruzi do not replicate within the bloodstream or tissues of the vertebrate host. Instead the trypomastigotes invade a tissue cell and transform into the amastigote form (step 2). T. cruzi is capable of invading many different cell types, but the spleen, liver, lymph nodes, and muscle are the most commonly affected organs. Invasion involves the recruitment of host cell lysosomes to a site on the host cell plasma membrane adjacent to the trypomastigote. These lysosomes fuse with the plasma membrane and the trypomastigote enters into this forming parasitophorous vacuole. Lysosomes continue to fuse with the parasitophorous vacuole as parasite entry proceeds. This entry does not appear to involve and actin-mediated phagocytosis process. Instead, the recruitment of lysosomes appears to be part of a process by which cells repair lesions on the plasma membrane. This parasitophorous vacuole is then disrupted and the trypomastigote escapes into the cytoplasm of the host cell. (Tan and Andrews, Tr. Parasitol., 2002, 18, 427.)
Once within the cytoplasm of the host cell the trypomastigote converts into the amastigote form. The amastigotes replicate by binary fission within the cytoplasm of the host cell and the progeny will fill up the cell. This intracellular cycle lasts an average of 4 days and after several rounds of division the amastigotes will differentiate into trypomastigotes and be released from the infected cell (step 3). These trypomastigotes can invade other host cells or enter the circulatory system. Trypomastigotes invading other host cells will transform back into amastigotes and repeat the replicative process. This asexual replication will continue until the vertebrate host is cured of the infection.
Trypomastigotes in the circulatory system can also be taken up by a triatomine bug during blood feeding (step 4). Within the vector's midgut the parasite differentiates to an epimastigote (step 5). The epimastigote will undergo multiple rounds of binary fission as it passes down the triatomine gut and attach to the epithelium of the rectal gland. Epimastigotes can also be grown in vitro. The epimastigotes quit dividing and differentiate back into trypomastigotes. After maturating to the infective metacyclic trypomastigote, the parasite detaches from the epithelial cells and remains in the lumen of the rectum awaiting excretion (step 6). This transformation to the trypomastigote form represents a preadaptation for the vertebrate host. Epimastigotes are lysed by host complement and therefore non-infective for the vertebrate host.
| Modes of T. cruzi Transmission | |
|---|---|
| SOURCE | COMMENTS |
| Vector (>80%) | Natural biological transmission by triatomine bugs via infected feces. |
| Transfusion (~16%) | Especially prevalent in urban areas. Gentian violet treatment eliminates parasites in blood. |
| Congenital (~2%) | Occurs during any stage of the infection. Can result in premature labor, abortion, or neonatal death. |
| Other (<1%) | Ingestion of food contaminated with metacyclic trypomastigotes. Laboratory accidents. |
Vector transmission is not the only mechanism by which humans can become infected with T. cruzi (Table). As with other blood-borne pathogens it is also possible to acquire the disease through blood transfusion or organ transplants. The parasite can also be transmitted from mother to fetus, often with severe outcomes. The ability of the metacyclic trypomastigotes to invade mucous membranes poses problems for laboratory workers. In addition to 'needle sticks', transmission can occur through rubbing the eyes with contaminated hands. It is also possible to acquire the infection by ingesting material contaminated with the parasite. The metacyclic trypomastigote and penetrate the mucous membranes around the gums and in the oral cavity. Mammalian reservoirs often acquire the infection by eating infected triatomines. Biological vector transmission (primarily rural areas) and blood transfusions (primarily urban areas) are the two most prevalent modes of acquiring the disease. Contaminated blood supplies are a major problem in many endemic countries. In the U.S., blood donations are not accepted from persons who have lived in a Chagas' endemic area. Currently there is no approved test with acceptable levels of sensitivity and specificity for screening potential donors.
| |
|
|
|
The transmission of T. cruzi via contaminated bug feces contrasts with the transmission of African trypanosomes via the saliva. This 'hindgut' transmission is quite inefficient. After the infective metacyclics are expelled with the feces they still must find a portal of entry, such as the bite wound or mucosal surface, into the vertebrate host. In contrast, the transmission of parasites via the mouthparts of the vector tends to be extremely efficient since the infective stage is being directly deposited into the vertebrate host. Trypanosomes transmitted via the hindgut or mouthparts are sometimes referred to as stercoraria or salivaria, respectively. A large percentage of the vectors for stercorarian trypanosomes (eg., triatomines) are infected in nature, whereas very few of the vectors for salivarian trypanosomes (eg., tse-tse) are naturally infected. The differences in transmission efficiencies are apparently compensated for by differences in vector infection rates.
The low transmission efficiency for T. cruzi means that considerable contact between humans and the vector are necessary for infection to occur. Such extended contact is usually associated with colonization of human habitats by triatomine bugs. Colonization of human habitats is most often associated with rural poverty. Factors such as adobe walls, thatched roofs, animal stalls adjacent to the house, proximity to the sylvatic cycle contribute to disease transmission. Under such conditions the triatomine bugs will come out of their hiding places during the night and feed upon the inhabitants. It is this repeated feeding over long periods of time that allow for transmission despite the rather inefficient mechanism. As further evidence of the inefficiency, only some members of a family will be infected with T. cruzi despite the fact that all members live in a triatomine infested house.
In nature T. cruzi exists in a sylvatic cycle involving the triatomines and various mammals. Generally, no overt disease is observed in the mammalian hosts and a natural balance between the vector, parasite and host is maintained. Human encroachment into the forests and destruction of the natural habitats forces the triatomine and mammalian reservoirs to colonize human domiciles and thereby initiating a domestic cycle (See Box in Molecular Evolution). Furthermore, at least six distinct subtypes have been identified by molecular studies. These subtypes all exhibit distinct geographical and ecological distributions (Zingales et al, 2012, Infect Genetics Evol 12:240).
| Chagas Control |
|---|
|
Chagas' control measures should prevent or limit the contact between humans and the vector. For example, improving human dwellings (eg., plastering inside walls, replacing thatched roofs) so that they are not subject to triatomine infestation greatly impacts transmission. Chagas' can also be controlled health education activities which result in behavior changes such as moving animal stalls away from the house. Infested houses can be cleared of triatomines, or the infestation prevented, through the use of insecticides. DDT generally has little effect against triatomines. Since the late 70's synthetic pyrethroid compounds have replaced organochlorides and organophosphates as the perferred insecticide. Although more expensive, the pyrethoroids are applied at lower doses and are less toxic than agents such as malathion. Urban transmission can be prevented by treating donated blood with gentian violet.
The Southern Cone Initiative against Chagas’ disease was formalized between Argentina, Bolivia, Brazil, Chile, Paraguay and Uruguay in 1991. The goal of this initiative was to interrupt the transmission of T. cruzi by eliminating Triatoma infestans from domestic settings. The control efforts consisted of spraying all houses and peridomestic areas with pyrethroid formulations. Spraying uninfested houses prevents the triatomines simply from moving house to house during the campaign. Community support was also enlisted to monitor for the reappearance of triatomines and respraying houses if triatomines reappeared. In addition, blood donors were also screened to prevent transmission via blood transfusions. These efforts have resulted in the interruption of T. cruzi transmission in large areas of these countries. Disease prevalence through south and central America has been reduced from an estimated 16-18 million people in 1990 to less than 10 million in 2006. There has also been a progressive decline in Chagas’ disease associated hospitalizations. Two other initiatives have been formalized as a result of the success of the Southern Cone Initiative: the Andean Pact Initiative between Venezuela, Colombia, Ecuador and Peru and the Central American Initiative (including Mexico).
Triatomines and T. cruzi-infected animals are found in many parts of the U.S., yet only a few cases of autochthonous transmission have been reported. (See case report for sixth reported case of autochthonous transmission in the U.S., Emerg Infect Dis 14:605, 2007.) Some of this lack of transmission is due to differences in the vector species found in the U.S. The vectors in North America tend to be zoophillic and avoid human contact. In addition, they are 'late' defecaters and do not excrete feces while taking a blood meal. However, socio-economic factors probably play a greater role in the restriction of Chagas' disease to South and Central America. Well constructed houses will not become infested with the triatomine bugs and therefore the extensive vector-human contact necessary for transmission does not occur. Similarly, there will be less peridomestic transmission in the United States.
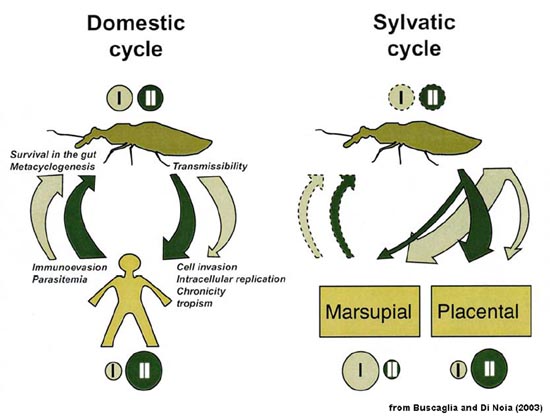
| Molecular Epidemiology and Evolution of Trypanosoma cruzi T. cruzi exhibits a high degree of polymorphism when analyzed by various biochemical and molecular techniques (1, 2). Despite this high level of polymorphism, two major phylogenetic groups are recognized regardless of the method used. However, there are a few parasite isolates that do not fit into either of these groups or may be hybrids. The two major groups have been designated as T. cruzi I (TcI) and II (TcII) in an effort to consolidate nomenclatures. There is even some discussion that TcI and TcII should be considered as distinct subspecies or species in that the genetic differences between them are greater than the differences between some Leishmania species. TcI and TcII also exhibit different biological and epidemiological features. For example, there is a preferential association of TcII with placental mammals, whereas TcI exhibits a preference for marsupials. Human infections are predominantly TcII with relatively few TcI infections. Furthermore, the TcI infections are more likely to be diagnosed during the acute stage, suggesting that TcI parasites may be cleared by the human host and not develop into a long-term chronic disease. In keeping with this higher prevalence of TcII in human infections, the TcII is associated with domestic and peridomestic transmission cycle, whereas TcI is associated with sylvatic cycles. This association of TcII with the domestic cycle cannot be solely attributed to a preference for triatomine species associated with domestic\peridomestic cycles in that the vectors exhibit a similar preference of infection for TcI and TcII and appear capable of transmitting both. The figure below summarizes these ecological (domestic and sylvatic cycles), epidemiological (scaled circles), and biological features (width of arrows) for TcI (light green) and TcII (dark green). (Dashed lines indicate that insufficient experimental data are available.)
In regards to its evolutionary history, T. cruzi is estimated to have emerged as a species more than 150 million years ago (mya). Phylogenetic analysis suggests that TcI and TcII diverged between 37 and 88 mya (3). This is a similar time period as the geographical isolation of North and South America. In other words, the split of Gondwanaland occurring approximately 100 mya led to the divergence of the two T. cruzi groups. The TcI type parasites originated in what would become North America and were primarily associated with marsupials, whereas the TcII type parasites were associated with placental mammals in South America. The continuous land connection at the Panama Isthmus, which formed 2-5 mya, led to an exchange of mammals and thus a geographical mixing of the two types. Although, both types are found throughout North and South America, TcI appears to be more predominant in the northern part of South America and northward, whereas TcII tends to be more predominant in the southern half of South America. Obviously, human contact with T. cruzi and the establishment of the domestic cycle are much more recent than the divergence of the two phylogenetic groups. Humans have been present in south and central America for at least 15,000 years and perhaps for as long as 20-25 thousand years. As humans came into contact with the natural sylvatic cycle, the triatomine vectors adapted to human habitations and allowed for the establishment of domicillary and peridomicillary transmission cyles. T. cruzi kinetoplastid DNA has been extracted from 4000 year old mummies buried along the northern coast of Chile (4). The higher prevalence of TcII infection and associated disease in humans may reflect the early evolution of TcII in placental mammals.
|
| |
|
|
|
| Phases of Chagas' |
|---|
acute phase
|
Chagas' disease is an extraordinarily complex process with a poorly understood pathophysiology. The disease exhibits three phases: acute, indeterminate (or latent), and chronic.
The acute disease is characterized by active infection with circulating trypomastigotes in the blood. Most persons are asymptomatic, or only have mild symptoms, and are unaware of having an acute illness. Children are more prone to develop recognizable symptoms and tend to have higher parasitemias. A hallmark of acute Chagas' disease is the Romaña sign, characterized by periorbital edema and conjunctivitis. An inflammatory lesion in the skin at the parasite entry site, known as a chagoma, develops less frequently. In both cases, these lesions represent the entry site for the parasite—either the mucous membranes around the eye or the bite wound--and a localized replication of the parasite as intracellular amastigotes. Lysis of the infected host cells results in the localized inflammation. Generally these inflammatory lesions appear a few days after infection and gradually subside over 2-3 months.
Clinical symptoms of the acute phase may also include: fever, malaise, lymphadenopathy, hepatosplenomegaly, vomiting, diarrhea. These symptoms spontaneously resolve within 3-4 months. A small number of individuals with develop and die of complications associated with an acute myocarditis or meningoencephalitis. Necropsy reveals extensive tissue pathology correlated with numerous amastigotes detected cardiac, skeletal and smooth muscle as well as glial cells.
Following the acute phase is an extended period of seropositivity in the absence of detectable parasitemia and clinical symptoms. This indeterminate or latent stage can last for decades and probably persists for the life of the infected person. During this latent period higher levels of electrocardiogram (ECG) abnormalities have been observed in seropositive persons than in seronegative persons. A right bundle branch block (RBBB) and left anterior hemiblock (LAHB) are the most notable abnormalities.
Chronic Chagas' disease most often manifests itself as a cardiomyopathy or autonomic neuronal dysfunction resulting in megasyndromes. Myocardial involvement in chronic Chagas' disease includes arrhythmias, conduction defects, cardiomegaly, congestive heart failure, and thromboembolic events. All parts of the heart (i.e., endocardium, myocardium, pericardium) can be can be affected. Heart disease is frequently associated with characteristic conduction disturbances, such as a complete atrioventricular block or a right bundle branch block. Chagas' cardiomyopathy is a progressive and relentless destruction of the myocardium and conduction system as suggested by the ECG defects. Progressive enlargement of the heart, congestive heart failure, and blood clots are other possible manifestations associated with Chagas’ disease. The micropathology reveals widespread areas of cellular infiltration and inflammation. Focal and diffuse areas of myocellular hypertrophy may also be seen with or without inflammatory infiltrates. Extensive fibrosis associated with the replacement of previously damaged myocardial tissue is also observed. The cellular hypertrophy and fibrosis correlate best with the cardiac symptoms and death is usually the result of rhythm disturbances or congestive heart failure. (For review see: ML Higuchi, LA Benvenuti, MM Reis and M Metzger. 2003. Pathophysiology of the heart in Chagas’ disease: current status and new developments. Cardiovascular Research 60, 96.)
| Megaviscerae |
|---|
|
Megasyndromes are another clinical manisfestation of chronic Chagas' disease. The prevalence of megasyndromes as compared to cardiac symptoms varies according to geographical location. Presumably T. cruzi isolates have a predilection to induce either megaviscerae or cardiomyopathy. The hollow viscera, especially the colon and esophagus, are most often affected. The dilation of these organs is possibly due to autonomic neuronal dysfunction. Upon autopsy a reduction in the number of ganglia associated with the colon and the esophagus has been noted in patients exhibiting megacolan and megaesophagus, respectively.
| |
|
|
|
| Basis of Pathogenesis |
|---|
| parasite-mediated destruction?
autoimmunity?
altered immune response?
|
The pathogenesis in chronic Chagas' disease is associated with inflammatory damage of the heart or with loss of parasympathetic nerves in the digestive system. Although it is clear that the disease is due to immunological attack of host tissues, the mechanisms are not completely known. The slow development of chronic stage and the lack of good animal models impede experimental studies.
Currently the most accepted theory is that the cardiac damage is due to an immune response elicited by persistent parasitemia. Histological analysis of tissues from chronically infected hosts reveals inflammatory cells or fibrosis with few or no parasites. However, parasites are routinely detected when more sensitive techniques, such as immunohistochemistry or PCR, are used and tissue damage is correlated with the presence of parasites. Therefore, the inflammatory response associated with chronic Chagas' disease appears to be due to the presence of parasites. In addition, in some studies treatment of chronic Chagas' disease lessened the clinical disease. An emerging picture is that T. cruzi persists in the host and results in a progressive disease with foci of inflammation and damage. Eventually the resulting damage reaches a clinically relevant level and produces the symptoms of cardiomyopathy or megasyndromes.The observation that chronic chagasic cardiomyopathy is generally accompanied by few, if any, parasites led to a prevailing dogma that the pathogenesis has an autoimmune or indirect etiology. The delayed onset of the disease and organ specificity are consistent with an autoimmune etiology. In addition, anti-self antibodies and lymphocytes have been observed during the course of infection. Mechanisms for generating autoimmunity could include molecular mimicry between parasite and host antigens (eg., similar antigenic epitopes) or bystander activation (eg., tissue destruction releasing autoantigens). However, auto-reactive antibodies (eg., anti-myosin) and immune effector cells are seen in other non-chagasic cardiomyopathies. Therefore it is not clear whether the 'autoimmunity' is the cause or the effect. In addition, immunosuppression tends to exacerbate rather than alleviate the disease indicating that autoimmunity does not completely explain the pathogenesis.
Others have proposed that an altered immune responses and/or toxic factors may play a role in the pathology. For example, the balance of Th1 and Th2 cells correlates with cardiomyopathy and disease severity. These various hypotheses are not mutually exclusive in that the disease may result from several factors. The fact that 30% of infected individuals develop clinical disease further emphasizes the incomplete understanding of the basis of pathogenesis of Chagas' disease.
(For reviews and more on the controversy, see R.L. Tarleton, Parasite persistence in the aetiology of Chagas disease, Int. J. Parasitol. 31:550, 2001; J.S. Leon and D.M. Engman, Autoimmunity in Chagas heart disease, Int. J. Parasitol. 31:555, 2001; and N. Girones and M. Fresno, Etiology of Chagas disease myocarditis: autoimmunity, parasite persistence, or both?, Tr. Parasitol. 19:19, 2003; DosReis et al, The importance of aberrant T-cell responses in Chagas disease, Tr. Parasitol. 21:237, 2005.)
| |
|
|
|
| Diagnosis |
|---|
clinical
|
Chagas' disease is suspected in persons exhibiting symptoms compatible with Chagas' disease (see above) and having an epidemiological history of possible exposure (ie, residence in substandard housing or receipt of unscreened blood products from an endemic areas). X-rays and other imaging techniques can be also used to detect the enlargement of the affected organs during the chronic stage. Confirmation is through laboratory diagnosis. The simplest methods are to examine fresh blood for motile trypomastigotes or Giemsa-stained thin and/or thick blood smears. Because of low parasitemias it may be necessary to employ concentration techniques such as examination of the buffy coat. Direct detection of the parasite is generally only possible during the acute stage. Other methods to increase the sensitivity include: inoculation of mice, in vitro culture, and xenodiagnosis. Xenodiagnosis is performed by allowing uninfected laboratory-raised triatomine bugs to take a blood meal from the patient and examining the bugs for infection. In vitro culture or xenodiagnosis will be positive in almost all acute cases and up to 50% of the chronically infected patients. Recently, PCR (polymerase chain reaction) has been used as a sensitive method for the detection of T. cruzi. Various serological tests are also available for the diagnosis of chronic stage Chagas' disease.
The two approved drugs for the treatment of Chagas' disease are nifurtimox (Lampit®, Bayer) and benznidazole (Rochagan® or Radanil®, Roche). Both drugs are believed to act primarily on the blood stream trypomastigotes and suppress parasitemia. The mechanism of action of nifurtimox is believed to involve the generation of reactive oxygen intermediates (i.e., superoxide and hydrogen peroxide), whereas benznidazole seems to act via a reductive stress mechanism. Parasitological cures of up to 80% have been obtained with treatment of either drug during the acute phase of the disease. However, the effectiveness of these drugs during the chronic stage is controversial. Some studies have shown a benefit to treating indeterminant or chronic stage patients (see Viotta et al 1994 for an example), whereas other studies have found no benefit to treating chronic stage patients (see Lauria-Pires et al 2000 for an example).
|
|
In addition, both drugs cause relatively severe and frequent side-effects, probably as a consequence of oxidative or reductive damage to host tissues. Thus many patients are unable to complete the long treatment schedules and many physicians have strong reservations about the use of nifurtimox and benznidazole during the chronic stage due to the questionable risk-to-benefit profile of these drugs. Alternative drugs are clearly needed, but few are currently under development. Although controversial, a set of guidelines for the treatment of Chagas' disease patients has been recommended by a panel of experts (see Table).
| Treatment Guidelines | ||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
|
Urbino, JA and Docampo R (2003) Specific chemotherapy of Chagas disease: controversies and advances. Tr. Parasitol. 19, 495.
Supportive treatment directed at relieving the symptoms may also be necessary in Chagas’ disease. Bed rest, hydration and antipyretics may be prescribed during symptomatic acute infections. In the case of heart failure during the chronic stage the supportive care is the same as for other forms of heart failure. Such care can include bed rest, diuretics, small doses of digoxin and possibly anticoagulants. Patients can also benefit from supervised physical activity. Anti-arrhythmic agents can be used to manage symptomatic or life-threatening arrhythmias. Pacemakers are also indicated in some situations. Mild symptoms associated with megaesophagus may respond to dietary changes or drugs to relax the lower esophageal sphincter. Surgical interventions may be prescribed in more advanced cases. Similarly, cases of megacolon which do not respond to diet, laxatives and enemas may require surgical intervention.
T. rangeli is another trypanosome with a geographical range that overlaps T. cruzi. It is also transmitted by triatomine bugs and infects a wide variety of mammals including humans. T. rangeli differs from T. cruzi in that metacyclic trypomastigotes are found in the salivary glands of the triatomine bug and is probably not transmitted via fecal contamination. T. rangeli is not pathogenic for the mammalian host and parasitemias tend to be quite low and transient. Interestingly, T. rangeli is quite pathogenic for the triatomine vector. The life cycle of T. rangeli in the mammal and vector is not completely known. Dividing trypomastigotes have been observed in mammalian blood and amastigotes have been observed in in vitro cultures. However, the significance of the these potential replicative forms is not clear. Similarly, the role of the various morphological forms found in the vector and how the parasite reaches the salivary glands is not clear. Because of the similar geographical ranges, vectors, hosts, and some serological cross-reactivity, it is possible to confuse T. rangeli and T. cruzi. However, the blood stage trypomastiotes are morphologically distinct. T. rangeli is a longer parasite with a smaller kinetoplast, similar to the African trypanosomes.
Reviews on Chagas Disease:
|
|
|
|
|
Several species of Leishmania are capable of infecting humans and causing disease. Leishmaniasis is expressed as a myriad of disease manifestations which depend upon parasite species, host responses, and poorly understood host-parasite-vector interactions. The different species and various clinical outcomes are also dependent on the geographic area and ecological setting. The number of reported cases and geographical areas in which leishmaniasis is endemic have been increasing.
| Leishmania Incidence |
|---|
|
Like other kinetoplastids, Leishmania are transmitted by a hematophagous arthropod. In this case it is a small biting fly known as a sand fly. Two different genera of sandflies transmit Leishmania: Phlebotomus (Old World) and Lutzomyia (New World). Sandflies are 'pool feeders' and promastigotes are transferred to the vertrebrate host during feeding. The promastigotes are not in the salivary glands, but found in the anterior portion of the gut and pharnyx. Infection of the sandfly leads to a breakdown in a valve at the entrance to the midgut. When the sandfly takes a blood meal the valve cannot close and blood moves back into the mouthparts and the bite wound. This regurgitation like activity results in an expulsion of promasitigotes from the anterior portions of the sandfly gut into the bite wound of the mammalian host.
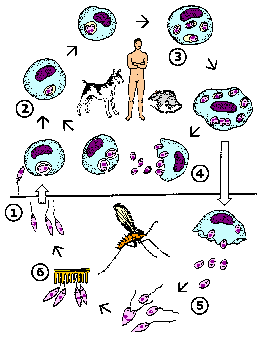 |
|
Leishmania life cycle (modified from K.P. Chang in Modern Parasite Biology edited by D.J. Wyler). See larger image. |
Within the vertebrate host the promastigotes are phagocytosed by macrophages or other 'professional phagocytes'. The parasite cannot actively invade host cells and is dependent upon the phagocytic activity of the host cell. Therefore, Leishmania is predominantly found in macrophage-like cells. This parasite-containing phagosome, as do other phagosomes, fuses with a lysosome. Normally a pathogen is destroyed in the resulting phagolysosome, but Leishmania is resistant to the acidic pH and hydrolytic enzymes present in the phagolysosome. In addition, the parasite shuts down the generation of reactive oxygen intermediates by the macrophage.
The promastigote converts to an amastigote within the phagolysosome by losing its flagellum and becoming more spherical. These amastigotes replicate by binary fission and fill up the infected macrophage. Amastigotes are release from the host cell and taken up by another macrophage leading to another round of replication. The release of the amastigotes is generally described as a bursting of the host cell. However, the parasitophorous vacuoles containing amastigotes have been observed to accumulate at the periphery of the infected cell and amastigotes are continuously released over several hours (Rittig et al, Infect. Immun. 66:4331; 1998). This suggests that amastigote release resembles an exocytosis-like process involving the fusion of the parasitophorous vacuolar membrane with the host cell plasma membrane.
Ingestion of infected macrophages by sandflies results in the conversion of the amastigotes to promastigotes. These procyclic promastigotes replicate by binary fission. They also attach to the midgut epithelium to avoid being excreted with the feces and continue to replicate. The development into metacyclic promastigotes is associated with a cessation of replication. This process is mimic during in vitro culture in that infectious promastigotes are predominantly found in non-dividing lag phase cultures. Changes in the biochemical properties of the promastigote surface also occur as the parasite matures to the metacyclic form. (For more discussion on Leishmania-sand fly interactions see vector page.)
|
|
|
|
|
Cutaneous leishmaniasis (CL) is the most common manifestation of the disease and is characterized by benign self-healing lesions that are generally painless and non-pruritic. The lesions can be ulcerated with a raised border or exhibit a papular or nodular nature (Figure). Quite often there is a progression from a papules, to nodules, and finally to ulcers. The ulcerated lesions can be classified as wet or dry depending on whether a crust forms over the lesion. Generally there is no pus associated with the wet lesions unless they become secondarily infected with bacteria. The lesion represents a localized infection at the site of the sandfly bite. The infection spreads outward from this point due to the continuing cycles of replication within the macrophages. The raised border of the lesion is where the active infection is occurring. Satellite lesions in the vicinity of the original lesion are sometimes observed. At some point the lesion will quit expanding and healing will start to take place. These lesions generally take weeks to months to heal. Scarring or pigmentation can be associated with the healed lesions.
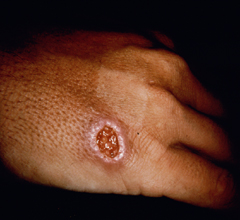 |
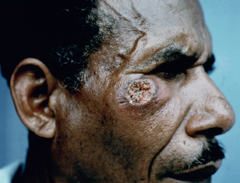 |
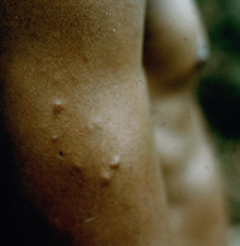 |
| Examples of cutaneous leishmaniasis. An ulcerated wet lesion (left), a dry lesion (middle), and satellite popular lesions surrounding the scar of a healed lesion (right). | ||
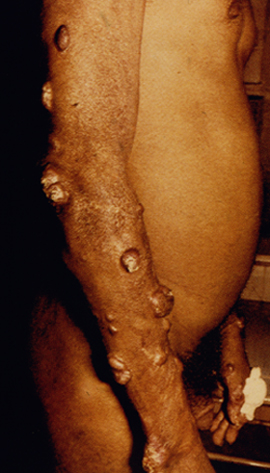
Diffuse cutaneous leishmaniasis (DCL) and leishmaniasis recidivans are two rare manifestations of cutaneous leishmaniasis. DCL is characterized by disseminated nodular lesions that resemble lepromatous leprosy. These lesions tend to be scaly and not ulcerated and can metastasize distally to cover large areas of the body. This condition is generally associated with anergy and amastigotes are abundant in the nodules. Recivida is a chronic recurrence of nodular lesions or a rash associated with L. tropica infections. The lesions tend to appear at the margin of the scar of the primary lesion. Few amastigotes are detected and this condition is considered to be a hypersensitivity in which most parasites are eliminated but the infection is not completely cured. Neither DCL nor recidiva is easily cured.
A small proportion (< 5%) of patients with simple cutaneous leishmaniasis will develop mucocutaneous leishmaniasis (MCL). This manifestation is primarily due to members of the L. braziliensis complex. Mucocutaneous disease begins as simple skin lesions that metastasize via the blood stream or lymphatics, particularly to the mucosae of the nose and mouth. The expression of this form of the disease can occur while the primary lesion is still active or can occur several years after the primary lesion has healed. Two distinct forms of MCL are recognized. One is an ulcerative disease with a rapid and extensive mutilation of soft tissue and cartilage. The other is non-ulcerative and characterized by local edema and hypertrophy, particularly of the upper lip. This disease will generally continue to progress and can lead to severe pathology and deformity if not treated.
 |
 |
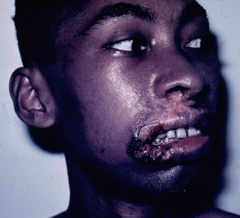  |
| Examples of recividans (left), diffuse cutaneous leishmaniasis (middle) and mucocutaneous leishmaniasis (right). | ||
Visceral leishmaniasis (VL) is the most serious form of the disease and can be fatal if untreated. It is a generalized infection of the reticuloendothelial system (RES) involving the spleen, liver, bone marrow and lymph nodes. The disease is characterized by fever, spleno- and hepatomegaly, and enlarged lymph nodes and tonsils. Patients often exhibit a wasting syndrome despite good appetite as the disease progresses. Other possible manifestations include edema, anemia, leucopenia, monocytoses, lymphocytoses, and thrombocytopenia. Inadequate treatment can lead to cutaneous disease known as post kala azar dermal leishmaniasis characterized by nodular lesions similar to DCL. In contrast to DCL, this post kala azar is easily cured with treatment.
Many Leishmania species are capable of infecting humans (see Table) and most are considered to be zoonoses. The species have defined geographical ranges and certain types of disease tend to exhibit a corresponding geographical distribution. Distinguishing Leishmania species based on morphological criteria at the light microscope is very difficult and quite often geographical and clinical criteria are used to assist in identifying species. Alternatively, immunological, molecular or biochemical criteria are needed to positively identify the species.
| Leishmania Species Infecting Humans | |||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
|
|||||||||||||||||||||||||||
| *Possibly the same species introduced to the New World by the European colonists. |
| L. chagasi = L. infantum? |
|---|
| Genetic analyses indicate the L. chagasi and L. infantum are closely related and some have proposed that they are the same species (Maurício et al, Parasitol. Today 16:188; 2000). However, the exact taxonomic status is currently being debated (Dantas-Torres, Mem Inst Oswaldo Cruz 101:929; 2006). Some data suggest that the parasite was introduced to the Americas with the arrival of the European colonists. Furthermore, L. infantum infects Lutzomyia as effectively as Phlebotomus indicating that a rapid transition to New World vectors is possible. |
Any Leishmania species is capable of causing any form of the disease. However, there is strong tendency for a particular species to exhibit a particular disease manifestation (Table). For example, in the New World L. mexicana infection tends to only cause simple CL, whereas L. braziliensis is much more likely to metastasize and result in a more serious MCL. Most notable is L. infantum which causes both cutaneous and visceral disease. Analysis of zymodemes (i.e., isoenzymes), however, indicates the presence of visceraltropic and dermotropic strains. Interestingly, dermotropic strains will frequently cause viseral disease in AIDS patients (see also discussion on L. infantum and AIDS). L. chagasi, which may be related to L. infantum (see box), is associated with a cutaneous disease characterized by non-ulcerated nodules as well as a visceral disease.
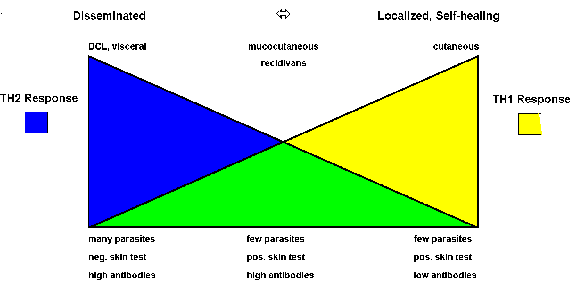
Both host and parasite factors are involved in disease outcome. Leishmaniasis can be viewed as a polar disease ranging from a lethal visceral disease to a self-curing cutaneous disease. Studies in rodent models indicate that the host immune response plays an important role in determining which of these extremes is expressed. Mouse strains resistant to Leishmania major mount a strong Th1, or cellular, response while susceptible mouse strains mount a predominant Th2 response.
The immunological aspects of human disease outcomes are more complex and less polarized than those in mice. However, there does tend to be a correlation with the differential expression of Th1 or Th2 immune responses (Figure). For example, localized self-healing cutaneous lesions have few parasites and the patient exhibits a positive Montenegro skin test and usually has low antibody levels. The skin test reflects a delayed hypersensitivity (i.e., Th1 response). On the other extreme is DCL and viseral leishmaniasis which are characterized by high parasitemias, a negative skin test, and high antibody titers (i.e., Th2 response). MCL and recidivans exhibit both Th1 and Th2 responses. These lesions are very slow to heal or fail to heal even though parasites are difficult to detect.
|
|
|
|
|
Diagnosis in endemic areas can be made based on clinical features, but does require considerable expertise. Definitive diagnosis depends on detecting the amasigotes in clinical specimens or culturing the promastigotes. The ability to detect parasites will depend on the abundance and distribution of amastigotes and whether the lesion is active or healing.
| Diagnosis of Leishmaniasis | |||
|---|---|---|---|
|
|||
|
|||
|
|||
|
|||
|
|||
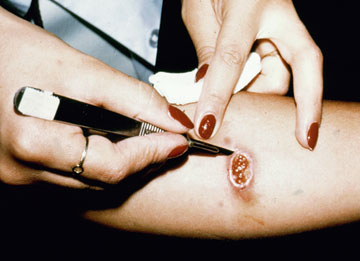
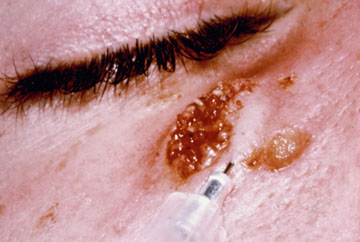
In the case of cutaneous leishmaniasis it is critical to collect the specimen from the border of the lesion where the infection is most active (Figure). An incision is made in the raised border of the ulcer and material scraped from the incision is examined microscopically. Alternatively, a biopsy can be taken from the lesion. The biopsy can be used to prepare impression or smears or sectioned. The lesion can also be aspirated by injecting sterile saline under the skin and withdrawing this fluid. The aspirate is then used to inoculate in vitro cultures. Inoculation of hamsters with the clinical specimens can be carried out, but may take 2-3 months before the animals become positive.
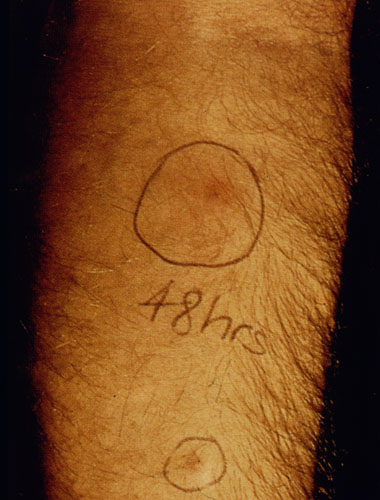
Another diagnostic test for cutaneous leishmaniasis is the Montenegro (leishmanin) skin test. Leishmania antigen is injected intradermally and monitored for a local delayed-type hypersensitivity reaction. The inability to distinguish current infections from past infections is a drawback to immunological methods.
 |
 |
 |
|
| Diagnosis of cutaneous leishmaniasis. (upper left) An incision along the raised border is made and dermal scrapings are examined by Giemsa staining and microscopy. (lower left) Sterile saline is injected into the raised border and fluid is aspirated. The aspirated can be examined microscopically or preferentially used to inoculate in vitro cultures or hamsters. Note the scar of the poorly positioned biopsy punch. Biopsies should be taken at the raised border of the lesion. (right) Montenegro delayed-type hypersensitivity reaction. | |
Examination of an aspirate or biopsy from bone marrow is the method of choice for diagnosis of visceral leishmaniasis. Aspirates or biopsies from lymph nodes or the spleen can also be used. Detection of amastigotes from visceral cases is generally easier than cutaneous cases since the parasitemias are higher in visceral leishmaniasis. The aspirates from microscopy negative samples can also be used to inoculate in vitro cultures to increase sensitivity. Serological tests are also available for the diagnosis of visceral leishmaniasis.
Anti-Leishmanial
Drugs |
|---|
|
Pentavalent antimonials are generally the first line of treatment. These drugs have been used for more than 50 years and are generally effective. However, they require parenteral administration and a long duration of therapy. Toxic side effects are also common. In addition, treatment failures have been observed over recent years suggesting the emergence of drug resistance.
Amphotericin B is an alternative to the pentavalent antimonials. It is effective, but does exhibit toxicity and also requires parenteral administration and a long duration of treatment. It is also more expensive than the pentavalent antimonials. Lipid formulations of amphotericin B have proven to be highly effective against visceral leishmaniasis with fewer toxic effects due to the requirement for lower doses. However, these formulations are prohibitively expensive for use in developing countries.
An oral drug for the treatment of visceral leishmaniasis has recently been registered in India. This new drug, miltefosine (Impavido®), is highly effective, less toxic, easily to administer, and will probably cost less than current therapies since hospitalization will not be required (see TDR news story). A phase IV clinical trial is now underway (see TDR news story).
| Key Epidemiological Features |
|---|
|
Leishmaniasis is endemic in tropical and sub-tropical regions throughout the world, as well as southern Europe. It is primarily a zoonotic disease with the exception of visceral leishmaniasis in India and some cutaneous leishmaniasis due to L. tropica. In general, the epidemiology of leishmaniasis will be reflective of the particular combination of parasite, vector and reservoir host of the local area (Box). Transmission in the New World is primarily associated with being near forests, whereas peridomicile transmission may predominate in the Old World. However, in many endemic areas the exact parasite, vector, reservoir, and their relations to human infections are not known.
Specific control activities will depend on the local transmission conditions. In general, sandfly bites should be avoided. Personal protection activities can include: protective clothing, insect repellants, bed nets, insecticide in domiciliary and peridomiciliary transmission settings. In some situations destruction of stray dogs and decreasing this potential reservoir has been effective in controlling leishmaniasis. However, in other situations elimination of dogs is not effective at controlling leishmaniasis. For example, elimination of infected dogs has not curtailed the spread of VL in Brazil. Costa et al (2002, AJTMH 66:334) showed a high level of asymptomatic carriers of L. chagasi in Brazil suggesting that humans may be a reservoir.
Historically vaccination by inducing the disease in a controlled manner has been practiced for the prevention of L. tropica. The procedure was to inoculate a covered area of the skin with living material obtained from an active lesion, or to encourage a sandfly bite in such an area. The resulting lesion spontaneously heals and prevents a lesion on the face or other exposed area. Although capable of providing lasting immunity, it is not recommended because of the risk of developing recidiva or visceral leishmaniasis. Efforts to develop a vaccine based on killed promastigotes are underway. Most studies indicate that vaccination does lead to conversion to leishmanin skin test positive, which is generally associated with a lower incidence of disease (see TDR News Story).
|
Leishmaniasis can be considered an emerging or re-emerging disease in that its geographical range is expanding and there appears to be an increasing incidence of disease. Factors underlying these changes are difficult to identify but may be related to global warming or changes in the behaviors of humans, vectors and reservoirs. Climatic changes may allow an extension of breeding seasons for existing vectors or the establishment of new vector species. For example, before 1990 northern Italy was almost free of sandflies and canine leishmaniasis. Since then the incidence of canine leishmaniasis has increased and several species of Phlebotomus are now found in northern Italy (1). In addition, there appears to be a northward expansion of L. infantum in other parts of Europe. Human factors affecting the endemicity of leishmaniasis include migration, deforestation, urbanization and an increase in susceptibility to infection due to immunosuppression (eg., AIDS) and malnutrition. Urbanization in South America is leading to an increase in leishmaniasis (2). Suburbs constructed on the outskirts of primary forests bring people into contact with the sylvatic cycle. In addition, these suburbs are often shanty towns where sandflies and dogs are common, sanitation is poor, and malnutrition is everywhere. Furthermore, the deforestation associated with this urbanization often leads to a domestication of the transmission cycle through adaptation of the vector and parasite to new reservoir hosts such as rodents and dogs. Accordingly, an increase in the peri- and intra-domiciliary transmission is being observed throughout Latin America, whereas previously, working in the forest was the primary risk factor. Similar increases in the suburban transmission of leishmaniasis in the Old World have also been noted.
|
Reviews on leishmaniasis:
These pages are developed and maintained by Mark F. Wiser, Tulane University (© 1999). Latest update February 8, 2022 .